Nederlandse Ziekenhuisfarmaciedagen 2023
- Rubriek: Congresabstracts
- Identificatie: 2023;8:a1774
Non-destructive drug content prediction of personalized 3D-printed tablets produced at the point-of-care using NIR and Raman spectroscopy
I. Lafeber a*, J.P. Bøtker b, D.M. Kweekel a, H.-J. Guchelaar a, J. Rantanen b and K.J.M. Schimmel a
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
b Department of Pharmacy, University of Copenhagen, Copenhagen, Denmark.
* Correspondence: i.lafeber@lumc.nl.
Background
Three-dimensional (3D) printing is a flexible production technique, enabling the production of tablets for individual patients at the point-of-care, such as in a hospital pharmacy. The quality of these tablets needs to be guaranteed, but conventional quality control is destructive and therefore not feasible. Due to the small batch sizes, non-destructive analytical methods are necessary. In this study, near-infrared (NIR) and Raman spectroscopy were assessed as non-destructive analytical methods for their suitability of predicting the drug content in personalized 3D-printed tablets produced at the point-of-care.
Methods
Tablets with diameters of 2.7 mm, 4.5 mm and 6.2 mm containing 10%, 15%, 20% and 25% w/w furosemide were produced at the hospital pharmacy of the Leiden University Medical Center using semi-solid extrusion 3D printing. Batch average concentrations were determined using high-performance liquid chromatography coupled with ultraviolet detection. Spectra of three tablets of each concentration and each size were measured using a tabletop NIR and a confocal Raman spectroscope. With NIR the tablets were analysed in triplicate from four sides. With Raman spectroscopy four, six or eight areas of 1 mm2 per tablet were analysed, depending on the tablet size, recording 100 spectra per area. The spectra were preprocessed and partial least squares (PLS) regression models were build using SIMCA software.
Results
Batch average concentrations (SD) were 10.89% (0.20%), 15.60% (0.39%), 20.21% (0.38%) and 25.19% (0.83%) w/w. Standard Normal Variate and data trimming was applied to the NIR spectra to acquire the optimal preliminary regression model (R2 = 0.9958; Q2 = 0.9948; RMSEE = 0.35%; RMSECV = 0.38%). For Raman spectral data, the spectra were averaged per area, cosmic rays removed and background removal applied to obtain the optimal preliminary regression model (R2 = 0.9799; Q2 = 0.9708; RMSEE = 0.77%; RMSECV = 0.96%). The current usability of Raman spectroscopy was assessed to be limited for use at the point-of-care, due to the long analysis time and potentially destructive properties of the laser.
Conclusion
Despite the small sample size, both preliminary models are sufficiently accurate and able to predict the furosemide concentration irrespective of the tablet size, indicating suitability of both NIR and Raman spectroscopy for the prediction of drug content in personalized 3D-printed tablets. NIR is favoured for use at the point-of-care, due to the limitations of Raman spectroscopy.
De abstractpresentatie van Iris Lafeber werd bekroond met de prijs voor Best Abstract 2023.
R-algorithm for identifying adverse drug reactions from free-text in electronic health records in hospitalized patients
Britt W.M. van de Burgt abc*, Arthur T.M. Wasylewicz b, Bjorn Dullemond d, Naomi T. Jessurun e, Rene J.E. Grouls a, R Arthur. Bouwman ch, Toine C.G. Egberts fg and Erik H.M. Korsten bc
a Division of Clinical Pharmacy, Catharina Hospital, Eindhoven.
b Division Healthcare Intelligence, Catharina Hospital, Eindhoven.
c Department of Electrical engineering, signal processing group, Technical University Eindhoven.
d Department of Mathematics and Computer Science, Technical University Eindhoven.
e Netherlands Pharmacovigilance Centre LAREB, ‘s-Hertogenbosch.
f Department of Clinical Pharmacy, University Medical Centre Utrecht.
g Department of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University.
h Department of Anesthesiology, Catharina Hospital, Eindhoven.
* Correspondence: britt.vd.burgt@catharinaziekenhuis.nl.
Objective
This study aims to develop a text mining tool using open-source R-algorithms to identify possible ADRs in free-text of Dutch hospital EHRs.
Methods
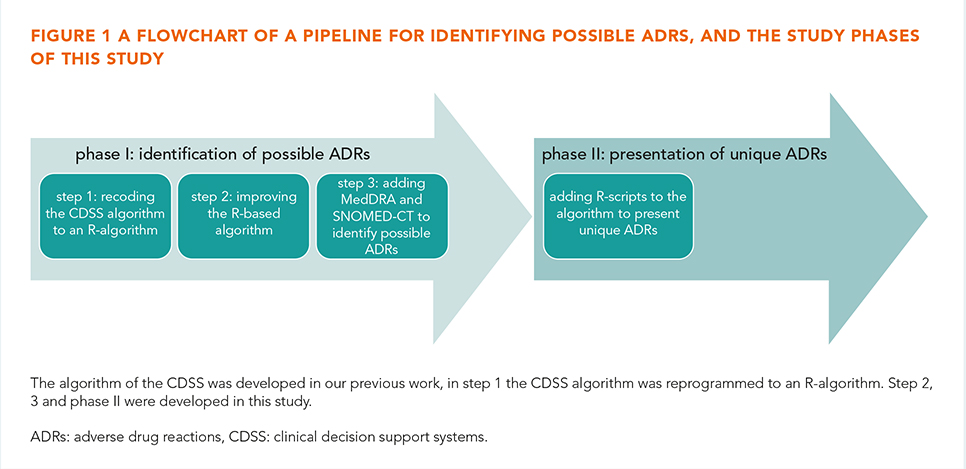
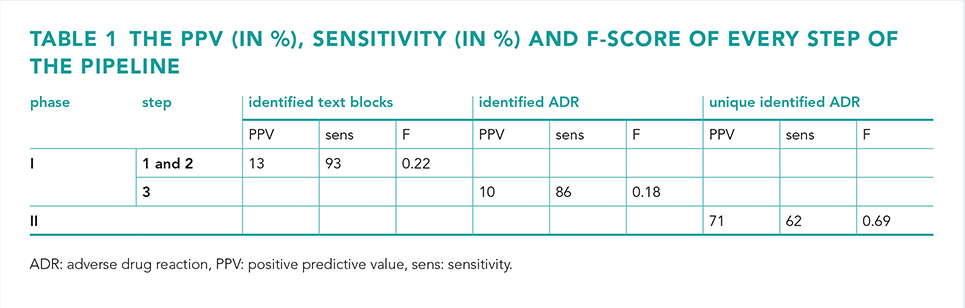
In our previous study, the complete EHR history of 45 patients were reviewed for ADRs and compared to two key strategies programmed into a CDSS (Gaston Pharma). ADRs were included in the study if the Naranjo causality score was ≥1. The defined gold standard found 318 unique EHR notes with possible ADRs, of which 63 potentially serious. In that study we demonstrated that a CDSS achieved a sensitivity of 57% and a positive predictive value (PPV) of 37% [6]. In the present study, in phase I the CDSS algorithm was recoded (step 1) and improved (step 2) using R to identify possible ADRs with MedDRA/SNOMED-CT (step 3). In phase II six existing text mining R-scripts (e.g. deduplication, negation and Levensthein distance) were studied to present unique ADRs and to improve the PPV and sensitivity (figure 1).
Results
In phase I step 1, the R-algorithm performed similarly to the GP algorithm. In step 2, the improved algorithm resulted in a 93% sensitivity and 13% PPV, with a sensitivity of 95% for potentially serious ADRs. The R-algorithm also identified an additional 58 possible ADRs. In step 3, the algorithm achieved a PPV of 10%, a sensitivity of 86% and an F-measure of 0.18. In phase II, four R-scripts enhanced the sensitivity and PPV, resulting in a 71% PPV, 62% sensitivity and 0.69 F-measure (table 1).
Conclusion
The R-algorithm effectively identifies ADRs from free-text Dutch EHRs using R-scripts and MedDRA/SNOMED-CT. Although the R-scripts perform significant better than the CDSS, there is still room for improvement before it can be integrated with a clinical decision support tool.
Real-world effectiveness versus clinical trial results of durvalumab in stage III unresectable non-small cell lung cancer
Hanieh Abedian Kalkhoran ab*, Loes E. Visser bcd, Egbert F. Smit e, Henk Codrington f, Henk-Jan Guchelaar a and Juliëtte Zwaveling a
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Centre.
b Department of Pharmacy, Haga Teaching Hospital, The Hague.
c Department of Hospital Pharmacy, Erasmus Medical Centre, Rotterdam.
d Department of Epidemiology, Erasmus Medical Centre, Rotterdam.
e Department of Pulmonary Disease, Leiden University Medical Centre.
f Department of Pulmonary Diseases - Pulmonic Oncology, Haga Teaching Hospital, The Hague.
* Correspondence: h.abedian_kalkhoran@lumc.nl.
Background
We compared the baseline characteristics and effectiveness outcomes of patients with stage III unresectable non-small cell lung cancer (NSCLC) who received durvalumab in a real-world (RW) setting with those from the PACIFIC trial.
Methods
In this retrospective study, we identified all patients diagnosed with stage III unresectable NSCLC who received durvalumab between April 2018 and February 2023 at two Dutch hospitals. We collected baseline and treatment-related characteristics, as well as effectiveness outcomes (overall survival [OS] and progression-free survival [PFS]) from the Electronic Health Record. These findings were then compared to the results of the PACIFIC trial. Patient selection and data collection were performed using a novel text-mining software tool (IQVIA Patient Finder Solution-CTcue B.V., Amsterdam, The Netherlands).
Results
Upon starting durvalumab treatment, RW patients exhibited less favorable prognostic indicators compared to the population in the PACIFIC study. Among those who received durvalumab in the RW setting, approximately 30% did not meet the eligibility criteria of the PACIFIC study. The median treatment duration with durvalumab was similar in both populations. However, the rate of treatment discontinuation due to immune-related adverse events was higher in the RW setting compared to the trial population (21% versus 15.4%). OS was similar between both populations (24-month OS: 69.3%; 95% confidence interval [CI] 59.3-80.9 versus 66.3%; 95% CI 61.7-70.4), while PFS was longer in the RW cohort (18-month PFS: 59.1%; 95% CI 49.4-70.7 versus 44.2%; 95% CI 37.7-50.5).
Conclusion
Despite the poor prognostic features of RW patients with stage III unresectable NSCLC, consolidation therapy with durvalumab resulted in a longer PFS than in the pivotal trial. OS was similar in both populations. The role of PD-L1 expression and prior chemoradiation therapy (sequential versus concurrent) as potential predictors for the effectiveness of durvalumab is currently being investigated in a follow-up study with a larger cohort.
Step-wise introduction of elexacaftor-tezacaftor-ivacaftor in patients with cystic fibrosis and liver cirrhosis Child-Pugh A or B using clinical and therapeutic drug monitoring: a case series
S.E.M. Vonk a, R. Lub b, E.J.M. Weersink b, U. Beuers c, R.A.A. Mathôt a, E.M. Kemper a* and J. Altenburg b on behalf of the Amsterdam Mucociliary Clearance Disease (AMCD) research group
a Amsterdam UMC location University of Amsterdam, Department of Pharmacy & Clinical Pharmacology.
b Amsterdam UMC location University of Amsterdam, Department of Respiratory Medicine.
c Amsterdam UMC location University of Amsterdam, Department of Gastroenterology and Hepatology.
* Correspondence: s.e.vonk@amsterdamumc.nl.
Background
Serum liver test abnormalities are described as a common adverse effect of elexacaftor-tezacaftor-ivacator (ETI) in patients with Cystic Fibrosis (pwCF). In the phase I registration studies the PK of ETI have been compared between non-CF people with hepatic impairment and healthy individuals. In the former group exposure of ETI was increased and therefore a reduced dose in pwCF and cirrhosis Child-Pugh B is recommended. To our knowledge there are no data on the exposure of ETI in pwCF and cirrhosis Child-Pugh A or B. In this case series we describe seven pwCF and cirrhosis Child-Pugh A or B where ETI was gradually introduced using clinical and therapeutic drug monitoring (TDM).
Methods
Four dosing steps were defined at which patients underwent clinical examination, routine blood tests and TDM. Exposure of ETI was assessed by determination of the area under the plasma concentration versus time curve (AUC). The decision to proceed with the next dosing step was at the discretion of the treating pulmonologist, taking into account the presence or absence of liver test abnormalities, other side effects, the TDM advice by the pharmacist and the clinical effectiveness of ETI.
Results
In all patients ETI was successfully introduced and maintained. All patients improved in respiratory symptoms, ppFEV1, and BMI. Four patients reported side effects, which resolved in most cases. In pwCF with Child-Pugh B cirrhosis (n = 2) diminishment of the dose as recommended by the label resulted in AUCs that were lower than previously reported mean AUC values in pwCF without hepatic impairment. Therefore, the dose was further increased under careful monitoring.
Conclusion
Stepwise elevation of ETI dose did not induce clinical side effects or increase in serum liver tests under strict clinical and biochemical follow-up and TDM, and may allow safe introduction of this therapy in pwCF and hepatic impairment.
Modifying tacrolimus-related toxicity after liver transplantation by using LCP-tacrolimus: a multicentre randomized, controlled trial (MOTTO)
M.B. Mulder ai*, B. van Hoek b, W.G. Polak ci, I.P.J. Alwayn d, B.C.M. de Winter ai, S. Darwish Murad hi, E. Verhey-Hart hi, L. Elshove hi, A. van den Burg hi, N.S. Erler Dipl.-Stat ef, D.A. Hesselink gi, C.M. den Hoed hi and H.J. Metselaar hi
a Department of Hospital Pharmacy, Erasmus MC, University Medical Center Rotterdam.
b Department of Gastroenterology and Hepatology, Leiden University Medical Center.
c Department of Surgery, Division of HPB and Transplant Surgery, Erasmus MC, University Medical Center Rotterdam.
d Department of Surgery, Leiden University Medical Center.
e Department of Biostatistics, Erasmus MC, University Medical Center Rotterdam.
f Department of Epidemiology, Erasmus MC, University Medical Center Rotterdam.
g Department of Internal Medicine, Division of Nephrology and Transplantation, Erasmus MC, University Medical Center Rotterdam.
h Department of Gastroenterology and Hepatology, Erasmus MC, University Medical Center Rotterdam.
i Erasmus MC Transplant Institute, University Medical Center Rotterdam.
* Correspondence: midas.mulder@haaglandenmc.nl.
Objective
The aim of this study was to investigate whether LCP-tacrolimus (Envarsus) compared to extended-release (ER)-tacrolimus (Advagraf) formulation results in a difference in the prevalence of post-transplant diabetes mellitus (PTDM), hypertension and chronic kidney disease (CKD) at 12 months after liver transplantation.
Methods
In this multicentre randomized, controlled trial, patients were randomized at discharge after liver transplantation (LT) in a 1:1 ratio to (1) ER-tacrolimus or (2) LCP-tacrolimus. The primary endpoint was a composite endpoint of any of three events at 12 months: CKD defined as eGFR < 60 mL/minute/1.73 m2 for > 3 months, sustained (> 3 months post LT) PTDM or new-onset hypertension. Secondary endpoints included: safety, quality of life, neurotoxicity (tremors), graft and patient survival, rejection, liver steatosis and fibrosis, pharmacokinetics and -dynamics.
Results
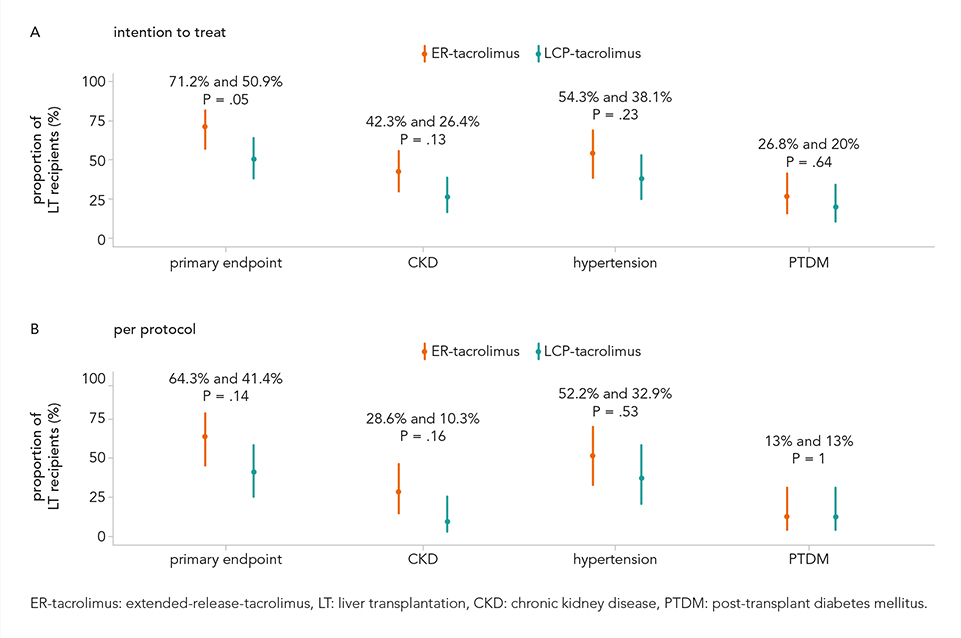
A total of 106 patients were included and baseline characteristics were comparable for both groups. In the intention-to-treat analysis, significantly less LT recipients reached the primary endpoint at 12 months in the interventional group compared to the control group (50.9% versus 71.2%, P = 0.05). The risk difference for the primary endpoint: 0.2021 and 95% confidence interval 0.002415-0.3816. In the intention-to-treat population, fewer LT recipients in the LCP-tacrolimus group developed CKD and new-onset hypertension compared to the ER-tacrolimus group: CKD 26.4% and 42.3%, P = 0.13 and new-onset hypertension 54.3% and 38.1%, P = 0.23. No difference was shown between ER-tacrolimus and LCP-tacrolimus in the percentage of LT recipients developing PTDM. No significant differences were observed in the per protocol analysis (see figure). In total, 95.3% (101/106) of the LT recipients developed serious adverse events (SAEs, n = 160). SAEs most frequently reported: fever (23.1%), infections (10%) and cholangitis and bile duct obstruction (10%).
Conclusion
LCP-tacrolimus results in a significant reduction in the prevalence of clinical relevant outcomes as compared to ER-tacroimus in the first year after liver transplantation with comparable efficacy.
The effect of colchicine on INR in patients with chronic coronary disease using vitamin K antagonists
Jeroen P.A. Houwen a, Jochem Zwaan b, A.C.G. Egberts c, Arend Mosterd d, Aernoud T.L. Fiolet e and A. Lalmohamed f
a University Medical Center Utrecht.
b Apotheek de Hoefslag, Barendrecht.
c Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht, Netherlands, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
d Dutch Network for Cardiovascular Research (WCN), Utrecht, The Netherlands, Department of Cardiology, Meander Medi cal Center, Amersfoort.
e Dutch Network for Cardiovascular Research (WCN), Utrecht, The Netherlands, University Medical Center Utrecht.
f Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht, Netherlands, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
Background
Low-dose colchicine improves cardiovascular outcomes in patients with chronic coronary disease. Therefore, a significant increase in the use of colchicine in cardiovascular therapy is expected. Around 12% of these patients are simultaneously prescribed anticoagulant therapy of which 25% comprises a vitamin K antagonist (VKA). In vitro studies and case reports describe the possibility of a drug-drug interaction between colchicine and VKAs. This interaction could be attributed to the inhibition of CYP2C9, potentially leading to an increased risk of bleeding. This possible interaction remains to be investigated.
Objective
To examine International Normalized Ratio (INR) and Time within therapeutic range (TTR) with or without colchicine use in those taking VKAs.
Methods
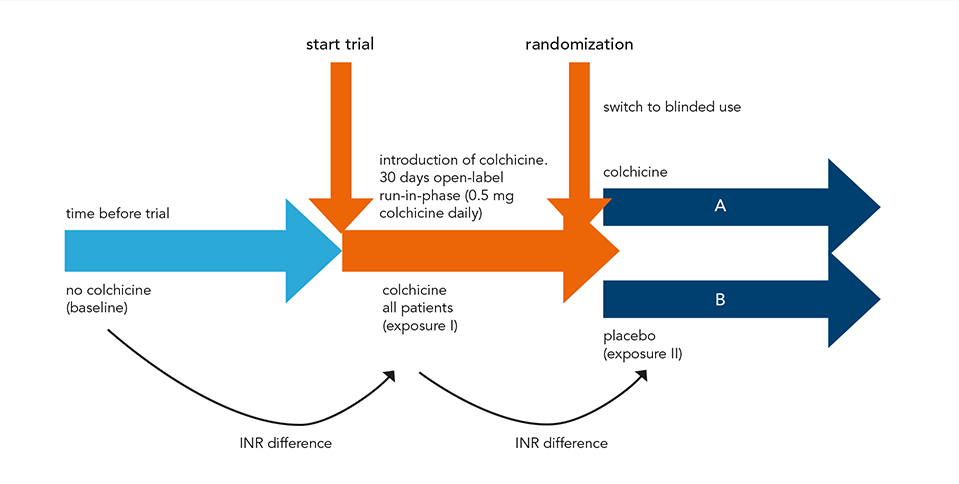
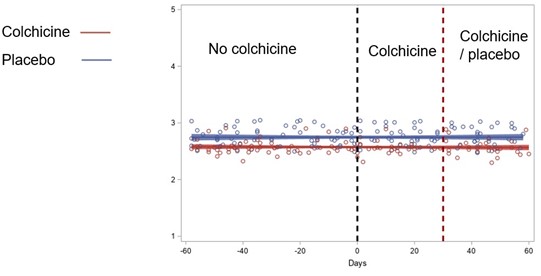
This study was a sub-analysis of the prospective, placebo-controlled LoDoCo2 trial. Patients with chronic coronary disease and concomitant VKA use were randomly assigned to receive either 0.5 mg of colchicine or placebo. Patients with INR data available from Dutch thrombosis services, were included. The primary outcome was the difference in TTR with colchicine versus placebo. Secondary outcomes included differences in INR after start and discontinuation, number of INR measurements, time between INR measurements and mean daily dosage of VKAs.
Results
A total of 82 patients were included in the study, with 39 assigned to the colchicine group and 43 to the placebo group. Baseline characteristics were similar between the two groups. The TTR difference (95% confidence interval [CI]) was 4.73% (−6.06%-15.52%) showing no significant change between colchicine and placebo users (P = 0.38) within 3 to 90 days after randomization. Overall TTR within this time period (95% CI) was 77.2% in the colchicine group (69.5%-84.9%) and 72.5% in the placebo group (65.0%-80.0%). These results remained consistent at 3 to 30 and 3 to 365 days after randomization. Similarly, the analysis of secondary outcomes, including INR difference after start and discontinuation, number of INR measurements, time between INR measurements and mean VKA daily dosage, revealed no significant differences.
Conclusion
In patients with chronic coronary disease receiving VKAs, low-dose colchicine did not significantly change TTR and INR compared to placebo. These results provide evidence for the safety of prescribing colchicine in patients using VKA, without the need for additional INR monitoring beyond standard care.
Incretin-based therapy and the risk of diabetic foot ulcers and related events
N.C.C. Werkman abc, J.H.M. Driessen abcd, O. Klungel b, N.C. Schaper ae, P.C. Souverein b, C.D.A. Stehouwer af and J.T.H. Nielen ac
a Cardiovascular Research Institute Maastricht (CARIM), Maastricht University.
b Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute of Pharmaceutical Sciences, Utrecht.
c Department of Clinical Pharmacy and Toxicology, Maastricht University Medical Center+, Maastricht.
d School for Nutrition and Translational Research in Metabolism (NUTRIM), Maastricht University.
e Department of Internal Medicine, Division of Endocrinology, Maastricht University Medical Center+.
f Department of Internal Medicine, Maastricht University Medical Center+.
Background
Incretin-based therapy in type 2 diabetes with dipeptidyl-peptidase-4 inhibitors (DPP4-Is) or glucagon-like peptide-1 receptor agonists (GLP1-RAs) has been associated with various beneficial effects beyond glycaemic control. Incretins might also have a positive effect on wound healing, which is important in the management of diabetic foot ulcers (DFUs), a severe complication in type 2 diabetes.
Objective
We aimed to investigate the effect of DPP4-Is and GLP1-RAs on DFU and DFU-related outcomes (lower limb amputation [LLA], DFU-related hospitalisation, and mortality).
Methods
We used data from the clinical practice research datalink (CPRD) Aurum database with linkage to hospital data and propensity score matched DPP4-I users to sulfonylurea users (n = 98,770), and GLP1-RA users to insulin users (n = 25,422). Cox proportional hazards models were used to estimate the hazard ratios (HRs) for DFU, LLA, hospitalisation, and mortality.
Results
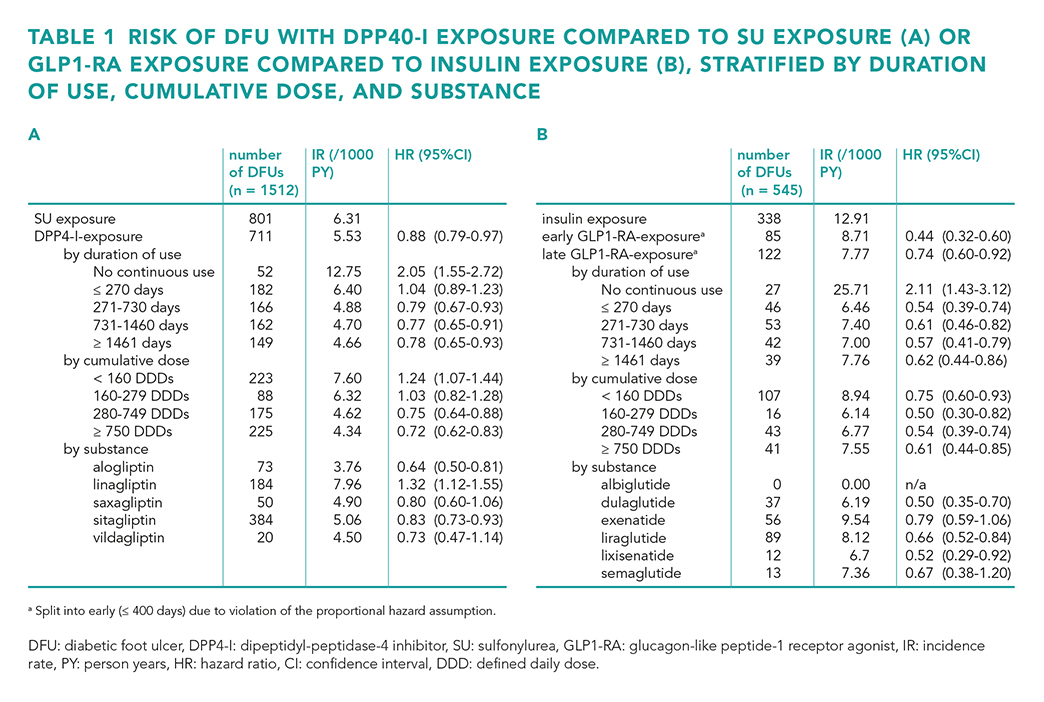
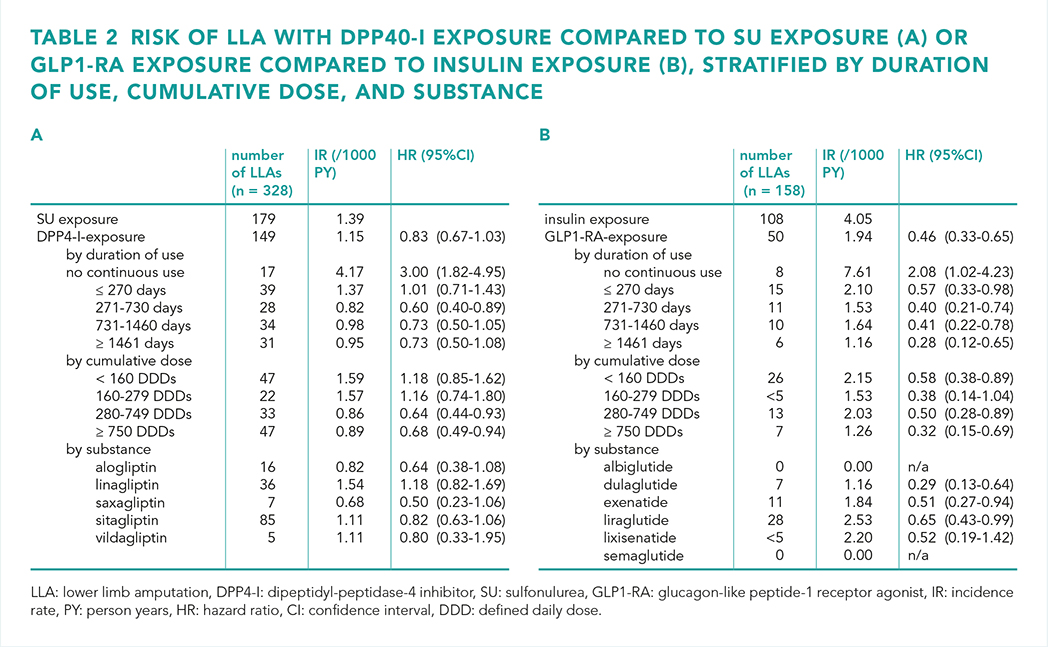
We observed a lower risk of DFU with both DPP4-I use versus sulfonylurea use (HR 0.88 (95% confidence interval [CI] 0.79-0.97) and GLP1-RA use versus insulin use (HR 0.44 (95% CI 0.32-0.60) for early exposure (≤ 400 days) and HR 0.74 (95% CI 0.60-0.92) for late exposure (> 400 days) (table 1). Furthermore, the risks of hospitalisation and mortality were lower with both DPP4-I use and GLP1-RA use. The risk of LLA was only lower with GLP1-RA use (table 2). The results remained consistent across a range of sensitivity analyses.
Conclusion
In this study, we observed a lower risk of DFU and DFU-related outcomes with incretin-based therapy. This might be an indication for the preferred use of this treatment type in people at risk of DFU.
Dose dependent crossreactivity of 15 insulin preparations across 8 different immunochemistry platforms: is fast toxicology screening possible?
C. Bethlehem a, I. Vleut a, J. Hillebrand b, S. Panchoe-Ramcharan a, M. Janssen c, J. Groen d, A. Muller-Kobold e, E. Lentjes f, A. Heijboer b, B. Koch a and S. Van Den Berg a
a Erasmus MC, Rotterdam.
b Amsterdam UMC.
c VieCurie Medisch Centrum, Venlo.
d IJsselland ziekenhuis, Capelle aan den IJssel.
e UMC Groningen.
f UMC Utrecht.
Background
In the differential diagnosis for emergency hypoglycemia, iatrogenic insulin is more common than metabolic disease or insulinoma. Laboratories rely on insulin measurements to establish the use of exogenous insulin preparations (EIP) [1]. However, not all insulin assays cross react with EIP [2] which may result in a false negative conclusion. Therefore, it is important to identify what EIP cross react on the local platform. Also, given the "need for speed" in the diagnosis, it may be possible to identify whether results of combinations of platforms may help in identification of the presence of EIP.
Methods
In a multicentre approach, 15 different insulin preparations were tested across 8 different immunochemistry platforms in a concentration (n = 7) dependent manner. In select cases of known EIP (as measured by LC/MS-MS), we compared the conclusion to the combination of an immunochemistry platform with and without cross reactivity.
Results
Cross reactivity patterns of all insulin preparations were established and compiled in a database. As expected, differences between platforms were found, with some platforms having no cross reactivity to non-isophane insulin and some platforms with cross reactivity to all insulin forms. In select cases, outcomes of LC/MS-MS were compared to immunoassay. EIP use was correctly identified.
Conclusion
We compiled a database of concentration dependent cross reactivity patterns for insulin preparations across 8 different immunochemistry platforms. Furthermore, we showed that a combination of results from immunochemistry platforms may be a promising fast method to identify exogenous insulin use.
References
1. Cryer PE, Axelrod L, Grossman AB, et al. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009 Mar;94(3):709-728.
2. Oh J, Kim JH, Park HD. Clinical Utility and Cross-Reactivity of Insulin and C-Peptide Assays by the Lumipulse G1200 System. Ann Lab Med, 2018 Nov;38(6):530-537.
Repurposing HLA genotype data of renal transplant patients to prevent severe drug hypersensitivity reactions
L.E.N. Manson a, S.J. Delwig a, J.J.M. Drabbels b, D.J. Touw c, A.P.J. de Vries de, D. Roelen be and H.J. Guchelaar a
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
b Department of Immunohematology, Leiden University Medical Center.
c Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
d Division of Nephrology, Department of Internal Medicine, Leiden University Medical Center.
e Leiden Transplant Center, Leiden University Medical Center.
Background and objective
From pharmacogenomic studies it is known that the risk alleles HLA-A*31:01, HLA-B*15:02, HLA-B*15:11, HLA-B*57:01 and HLA-B*58:01 are associated with an increased risk of developing drug hypersensitivity reactions induced by abacavir, allopurinol, carbamazepine, oxcarbazepine, phenytoin, lamotrigine or flucloxacillin. Pre-emptive genotyping is only introduced for abacavir-HLA-B*57:01. Since transplant patients are routinely genotyped for human leukocyte antigen (HLA) genes to assess whether donor and recipient can be matched, we aimed to investigate the feasibility of repurposing HLA genotyping results for the prevention of drug hypersensitivity reactions.
Methods
HLA genotyping by Next Generation Sequencing (NGS) is routinely performed in the Leiden University Medical Center (LUMC) for all transplant recipients and donors. The risk alleles HLA-A*31:01, HLA-B*15:02, HLA-B*15:11, HLA-B*57:01, and HLA-B*58:01 were retrieved from the NGS data. Medical history, medication use, and allergic reactions were obtained from the patient’s medical records.
Results
13.3% of transplant cohort patients carried at least one of the five HLA risk alleles and are therefore at risk for drug induced hypersensitivity. HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, and HLA-B*58:01 were found in carrier frequencies of 4.61%, 1.19%, 4.46%, and 3.35% respectively. No HLA-B*15:11 carrier was found. In total nine HLA-B*57:01 carriers had received flucloxacillin and seven HLA-B*58:01 carriers within our cohort received allopurinol. None of these patients developed a drug hypersensitivity reaction.
Conclusion
Repurposing HLA NGS genotyping results for the prevention of drug hypersensitivity reactions is feasible and leads to the detection of patients at risk for drug induced hypersensitivity. Although the positive predictive value of the HLA tests is low, due to the severity of the associated drug hypersensitivity reactions, documenting these risk alleles as a contraindication in the electronic health record, may prevent drug hypersensitivity reactions of transplant recipients.
Vancomycin flushing reaction after intraperitoneal vancomycin: a case report
J.E. Möhlmann a, A.M.K. Daza Zabaleta b, M. van Luin a and A.C Abrahams b
a Afdeling Apotheek, Universitair Medisch Centrum Utrecht.
b Afdeling Nefrologie en Hypertensie, Universitair Medisch Centrum Utrecht.
Vancomycin has been reported to cause vancomycin flushing reaction (VFR), a hypersensitivity reaction that mostly occurs after intravenous administration. The incidence of VFR in a patient receiving intraperitoneal vancomycin is rare. We report a case of a female peritoneal dialysis (PD) patient with a PD-related peritonitis who developed VFR after intraperitoneal administration of 2000 mg vancomycin. Seventy-five minutes after instillation, she developed flushing, a pruritic erythema on the upper body and swelling of the lips. Blood results revealed a vancomycin plasma concentration of 54.8 mg/L and a normal tryptase level. During a relapse of her PD-related peritonitis, vancomycin was successfully reintroduced in a 50% reduced dose. No symptoms of VFR developed and the corresponding vancomycin plasma concentration was 33.6 mg/L. Intraperitoneal treatment was continued with 500 mg vancomycin every 2-3 days with frequently measured, adequate trough levels ranging from 15 to 22 mg/L. This case illustrates the risk factors for the development of VFR after intraperitoneal administration of vancomycin, namely a high and concentrated loading dose together with a low body weight, a fast peritoneal transport state and peritonitis. Reintroduction of vancomycin after occurrence of VFR is safe, but a lower loading dose or a slower instillation rate is recommended.
Delabeling of penicillin allergies during medication reconciliation in hospitalized patients with reported antibiotic allergies
S.E.J.D. van den Eijnde, L.E. Gamadia and P.D. van der Linden
Tergooi Medical Centre, Hilversum.
Background and objective
Approximately 10% of hospitalized patients report having an antibiotic allergy, with penicillin allergies being the most prevalent. However, research shows that 90% of these patients are not truly allergic which leads to unnecessary avoiding of penicillin antibiotics. Alternative antibiotics have more adverse events, are less effective or increase the risk of antibiotic resistance development. We therefore aimed to develop a method to reduce the number of unnecessary antibiotic allergies and remove redundant penicillin allergies registrations in hospitalized patients.
Methods
Patients who were hospitalized, or had an elective operation planned, and reported an antibiotic allergy between July 2022 and June 2023 were included in this stewardship intervention study conducted at Tergooi Medical Centre. The control period was between June 2021 and May 2022. To assess the severity, a structured antibiotic questionnaire was filled in by pharmacist technicians during medication reconciliation. A recommendation based on an algorithm was provided in the electronic medical record. An allergy team, including an internist-allergist and hospital pharmacist, had weekly meetings to review the recommendations. The penicillin allergies could be directly removed, an oral provocation test was recommended or the patient was referred to the allergist. In addition, the days of antibiotic therapy (DOT) per 100 admissions were calculated.
Results
In the control period, 1394 antibiotic allergies were reported, of which 848 patients reported a penicillin allergy (60.9%). During this period, 156 penicillin allergies were removed (18.4%). During the intervention period, 1276 antibiotic allergies were reported. In addition, 876 questionnaires in 794 patients for a penicillin allergy were filled in. A total of 421 penicillin allergies were removed (53.0%) and 263 (62.5%) were directly removed due to the intervention. An oral provocation was advised in 226 cases, however only 36 provocation tests (15.9%) were performed and zero allergic reactions were observed. Overall, during the control period there were 248.0 DOTs/100 admissions, while in the intervention period, this increased to 252.5 DOTs/100 admissions. Additionally, an increase in penicillin use (68.9 versus 74.0) and a decrease in ciprofloxacin (18.5 versus 17.4) was observed in the intervention period.
Conclusion
This stewardship intervention demonstrated a method to improve the antibiotic allergy registration and to reduce the number of penicillin allergies labels in hospitalized patients. However, to successfully implement an oral provocation during hospitalization remains challenging due to the condition of the patient, short admission time and extra time investment of nurses and doctors in the hospital.
Physiologically-based pharmacokinetic modelling of methotrexate to predict exposure in adults and infants
P. Mian ab, C.H.T. Yeung c, M.S. Kim a, S. Ito c and A.N. Edginton a
a School of Pharmacy, Faculty of Science, University of Waterloo, Kitchener, Ontario, Canada.
b Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
c Division of Clinical Pharmacology and Toxicology, Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada.
Background and objective
Low-dose and high-dose methotrexate (MTX) are used to treat rheumatoid arthritis and malignancies, respectively. Little is known about the pharmacokinetics (PK) of MTX in special populations, such as infants. It is also speculated that MTX exposures occur in the infant breastfed by the mother who is treated with MTX. The objective of this study was to apply physiologically-based pharmacokinetic (PBPK) modelling to predict infant drug exposure based on drug monitoring data of MTX.
Methods
PBPK models for both low and high-dose MTX were developed using PK-Sim (Open Systems Pharmacology) in adults with rheumatoid artritis or malignancies. The anatomy and physiology –including maturation processes like metabolic capacity, glomerular filtration rate, protein binding and transporters – were scaled to that of a virtual population of 1000 infants between 0-12 months. Models were evaluated using goodness-of-fit plots and by comparing predicted PK profiles with in vivo PK data.
Results
The developed PBPK model successfully captured MTX plasma concentrations in infants. The ratios of prediction versus observation for MTX area under the curve from time 0 to infinity (AUC0-∞) and maximum concentration (Cmax) were 1.02 and 1.17, respectively. All predictions of AUC and Cmax for MTX were within a 1.25-fold error range.
Conclusion
These findings demonstrate the feasibility of translating PBPK models to infants with malignancies, as well as those breastfed by the mother on MTX, to potentially guide clinical decision making. Our next step will be to predict infant drug exposure of MTX through breast milk.
Is higher docetaxel clearance in prostate cancer patients explained by higher CYP3A activity? An in vivo phenotyping study with midazolam
L.T. van der Heijden a, C.A. Ribbers ab, M.A.C. Vermunt a, D. Pluim a, M. Acda a, M. Tibben a, H. Rosing a, J.A.J. Douma cd, K. Naipal c, A.M. Bergman ce, J.H. Beijnen afg, A.D.R. Huitema ahi and F.L. Opdam a
a Department of Pharmacy & Pharmacology, Antoni van Leeuwenhoek/The Netherlands Cancer Institute, Amsterdam.
b Department of Pharmaceutical Sciences, Utrecht University.
c Department of Clinical Pharmacology, Division of Medical Oncology, Antoni van Leeuwenhoek/The Netherlands Cancer Institute, Amsterdam.
d Department of Internal Medicine, Medisch Centrum Leeuwarden.
e Department of Oncogenomics, The Netherlands Cancer Institute, Amsterdam.
f Division of Pharmaco-epidemiology and Clinical Pharmacology, Faculty of Science, Department of Pharmaceutical Sciences, Utrecht University.
g Modra Pharmaceuticals B.V., Amsterdam.
h Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University.
i Department of Pharmacology, Princess Máxima Center, Utrecht.
Background
Docetaxel exposure is significantly lower in metastatic castration resistant prostate cancer (mCRPC) patients compared to patients with other solid tumours (OST) for intravenous (1.8-fold) and oral (2.4-fold) administration, which is associated with less neutropenia. This difference in docetaxel exposure is specific for prostate cancer (PC) and independent of disease status. Lower docetaxel exposure for PC patients is hypothesised to be caused by an increased cytochrome P450 3A (CYP3A) activity and/or increased hepatic uptake, resulting in an increased clearance.
Objective
The aim of the current study was to quantify in vivo CYP3A activity in PC patients and male OST patients, defined as midazolam clearance (validated metric for CYP3A activity). Secondary objectives were the comparison of midazolam area under the plasma concentration curve extrapolated to infinity (AUC∞) and the metabolic ratio to 1’-hydroxy midazolam between the two groups.
Methods
An in vivo phenotyping study was conducted, including 9 PC patients and 9 male OST patients. All male patients with solid tumours who did not use CYP3A modulating drugs were eligible for participation. Participants received 2 mg midazolam orally and 1 mg intravenously on two consecutive days. Non-compartmental analysis was performed to calculate midazolam clearance, AUC∞ and metabolic ratio. The following single nucleotide polymorphisms (SNPs) were determined: CYP3A4*2 (664T>C), CYP3A4*17 (566T>C), CYP3A4*22 (15389C>T) and CYP3A5*3 (6987A>G).
Results
Oral midazolam clearance was 1.26-fold higher in PC patients compared to male OST patients but this difference was not statistically significant (geometric mean (CV%): 94.1 (33.5%) L/h versus 74.4 (39.1%) L/h, respectively; P = 0.08). Intravenous midazolam clearance did not significantly differ between the two groups (P = 0.93). Oral AUC∞ was lower for PC patients compared to male OST patients (21.2 (33.5%) versus 26.9 (39.1%) ng/mL*h, respectively; P = 0.08), which was statistically insignificant, while intravenous AUC∞ was similar between the two groups (P = 0.93). Moreover, the metabolic ratio of midazolam to 1’-hydroxy midazolam did not differ between the two groups for both oral administration (P = 0.67) and intravenous administration (P = 0.26). CYP3A4 and CYP3A5 genotypes did not influence midazolam pharmacokinetics.
Conclusion
Oral midazolam clearance was 1.26-fold higher in PC patients compared to male OST patients, while intravenous midazolam clearance was similar between the two patient groups. The observed difference in oral midazolam clearance could not explain the previously reported difference in docetaxel exposure between the two patient groups. An alternative (but currently hypothetical) explanation for the difference in docetaxel pharmacokinetics could be the upregulation of hepatic transporters, increasing hepatic uptake and clearance of docetaxel.
Impact of bariatric surgery on oral anticancer drugs: an analysis of real-world data and practical recommendations
C. Lau abdg†, M.I.M. Mohmaed ab†, L. Lin ab, D.E.M. van Balen a, B.A.W. Jacobs a, B. Nuijen a, R.M. Smeenk e, N. Steeghs c and A.D.R. Huitema afg
a Department of Pharmacy and Pharmacology, Antoni van Leeuwenhoek Hospital/The Netherlands Cancer Institute, Amsterdam.
b Division of Pharmacology, The Netherlands Cancer Institute, Amsterdam.
c Division of Medical Oncology, Antoni van Leeuwenhoek Hospital/The Netherlands Cancer Institute, Amsterdam.
d Department of Clinical Pharmacy, Albert Schweitzer Hospital, Dordrecht.
e Department of Surgery, Albert Schweitzer Hospital, Dordrecht.
f Department of Pharmacology, Princess Máxima Center for Pediatric Oncology, Utrecht.
g Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University.
† These authors have contributed equally to this work and share first authorship.
Background and objective
The number of patients with a bariatric surgery who receive oral anticancer drugs is rising. The absorption of orally administered oncolytic drugs may be affected by bariatric surgery. However, no specific drug dosing advice is yet available for these patients. In this study, the prevalence of bariatric patients and the potential impact of bariatric surgery on the exposure to oral anticancer drugs were investigated. Based on this analysis, practical recommendations on the application of oral anticancer drugs were proposed for patients who underwent bariatric surgery.
Methods
The numbers of bariatric patients, the oral anticancer drugs that they used and measured drug levels in these patients were extracted retrospectively in a comprehensive cancer center. Based on the biopharmaceutical properties of the oral anticancer drugs, a decision tool was developed to assess the risk of underdosing of these drugs in patients who underwent bariatric surgery. With the decision tool, each drug that was prescribed to a bariatric patient was first categorized into low (no changes in pharmacokinetics expected), medium (Therapeutic Drug Monitoring [TDM] guided dosing recommended) or high risk (drug switch proposed). Subsequently, the decision tool was evaluated retrospectively using routine TDM samples.
Results
In total, 571 patients who underwent any kind of bariatric surgery in their medical history were included. In total, 78 unique patients received 152 oral anticancer drugs equaling an overall number of 30 unique drugs. Based on the decision tool, the 30 different prescribed oral oncolytic drugs were categorized as low risk (13%), medium risk (67%) and high risk (20%). 82 routine TDM plasma samples of 25 patients were available. Of the patients receiving medium risk oral oncolytic drugs, 47% needed a TDM guided dose increase in order to reach adequate drug exposure. One low-risk drug was identified. Patients treated with this low-risk drug had both adequate and inadequate exposure. Lastly, the exposure of three high-risk drugs remained low despite TDM guided dose interventions.
Conclusion
Bariatric patients treated with orally administered drugs deserve additional monitoring and may need to switch to other drugs with lower risks of underdosing. The proposed decision tool can support clinicians and pharmacists in making decisions regarding the optimal treatment with orally administered oncolytic drugs in patients who underwent bariatric surgery.
Retrospective analysis of deprescribing antithrombotic medication in palliative cancer patients with solid tumours in Amsterdam UMC
A.L. Riveras a, M. Crul a, J.M. van der Kloes a, M.A.H. Steegers b and B.A.A. Huisman b
a Apotheek en Klinische Farmacologie, Amsterdam UMC, locatie Vrije Universiteit.
b Anesthesiologie, Amsterdam UMC, locatie Vrije Universiteit.
Background and objective
Treating palliative cancer patients with antithrombotics is challenging because of the higher risk for both venous thromboembolism and major bleeding. There is a lack of available guidelines on deprescribing potentially inappropriate antithrombotics. Palliative medicine specialists from the Amsterdam UMC and Erasmus MC have therefore created an antithrombotics scheme to aid in (de)prescribing antithrombotics. This research aims to analyze the use of this scheme for antithrombotics retrospectively.
Methods
A retrospective single-centre clinical cohort observational study was performed in Amsterdam UMC. Palliative care patients seen in 2021 with solid tumours with a life expectancy of fewer than 3 months were included. Patients’ files were manually reviewed to retrospectively collect data on the (de)prescription of antithrombotics, reasons surrounding (de)prescribing antithrombotics, and the occurrence of clinical events. Comparisons were made between the group with scheme adherence and the group without scheme adherence.
Results
152 of 319 (47.6%) patients seen in 2021 by the palliative care team used antithrombotics. Of these, 111 were included in this research. Most patients used antithrombotics according to the antithrombotics scheme (n = 80, 72.1%). 11 patients experienced a clinical event, 7 patients in the scheme adherence group (9.9%) and 4 patients in the no scheme adherence group (13.8%), which was not statistically significant (P = 0.726).
Conclusion
In conclusion, the higher frequency of clinical events in the group without scheme adherence suggests that (de)prescribing antithrombotics according to the antithrombotics scheme is safe. The antithrombotics scheme could be beneficial to patients and it aids healthcare professionals in identifying possible inappropriate antithrombotics. Prospective research is needed to validate the antithrombotics scheme.
Relevance of MIR27A polymorphisms in relation to 5-FU associated toxicity in patients receiving DPYD genotype-guided dosing
S.L.J. Peeters ai, Zerina Kadric a, Didier Meulendijks b, Sylvie J. Kolfschoten-van der Kruijs c, Birgit A.L.M. Deiman d, Sara Ibrovic a, Vanja Milosevic e, Matthijs van de Poll f, Lieke H.J. Simkens g, Geert-Jan Creemers h and Maarten J. Deenen ai
a Department of Clinical Pharmacy, Catharina Hospital, Eindhoven.
b Late Development Oncology, AstraZeneca, Cambridge, United Kingdom,
c Department of Education and Research, Catharina Hospital, Eindhoven.
d Department of Molecular Biology, Catharina Hospital, Eindhoven.
e Department of Clinical Pharmacy, Elkerliek Hospital, Helmond.
f Department of Clinical Pharmacy, Máxima Medical Centre, Eindhoven.
g Department of Medical Oncology, Máxima Medical Centre, Eindhoven.
h Department of Medical Oncology, Catharina Hospital, Eindhoven.
i Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
Background
DPYD variant alleles (*2A, *13, c.1236G>A, and c.2846A>T) have been established as predictors of 5-FU associated toxicity and carriers are now treated with a genotype-guided dose of 5-FU. However, there is a variability between the DPYD genotype and DPD phenotype, with some DPYD variant allele carriers tolerating up to standard doses of 5-FU without major toxicity. Identification of novel factors contributing to interindividual variability may enable more effective patient stratification for 5-FU toxicity. The expression of DPYD has previously been shown to be regulated by miR-27a and a common MIR27A variant (rs895819) has been associated with increased risk of 5-FU toxicity in patients that harbor a DPYD variant allele. Therefore we investigated if MIR27A polymorphism was associated with 5-FU toxicity in DPYD variant allele carriers following DPYD genotype-guided dosing.
Methods
This was a retrospective, multicentre, pharmacogenetic association study conducted in three hospitals in the Netherlands. 95 heterozygous DPYD variant allele carriers treated with DPYD genotype-guided dosing of 5-FU were identified. Patients were genotyped for MIR27A rs895819 from prior obtained DNA. Associations of MIR27A polymorphisms with early-onset (cycles 1-3) 5-FU toxicity and relative dose intensity were assessed.
Results
52 patients had a MIR27A variant allele and 43 were wildtype. MIR27A variant rs895819 did not result in significantly increased risk of severe ≥ grade 3 toxicity compared to MIR27A wild-types in heterozygous DPYD*2A, DPYD*13, c.1236G>A, and c.2846A>T carriers. Relative dose intensity was found to be the strongest predictor of 5-FU toxicity in multivariable logistic regression models. The major finding of our study was that the median relative dose intensity of heterozygous c.2846A>T variant allele carriers who had an initial dose around 75%, was significantly reduced from 74% in cycle 1 to 66% in cycle 3 due to toxicity (P = 0.03). The median relative dose intensity of heterozygous c.1236G>A variant allele carriers who had an initial dose around 75%, was only significantly reduced from 75% in cycle 1 to 72% in cycle 3 due to toxicity (P = 0.03). In addition, we observed oncologists did not escalate 5-FU dosage in a proportion of patients without ≥ grade 3 toxicity.
Conclusion
This study did not find an association between MIR27A polymorphisms and 5-FU toxicity, which may be explained by the lower toxicity risk of this patient group because of DPYD genotype-guided dosing. The results indicate c.1236G>A and c.2846A>T can tolerate higher 5-FU doses (75% and 65% respectively) and uptitration in these patients should be considered. Future studies regarding 5-FU dose escalations and MIR27A are warranted.
Excessive duration of antibiotic therapy in patients with common infections in Tergooi MC
S.E.J.D. van den Eijnde, T.M. Baars and P.D. van der Linden
Tergooi Medical Centre, Hilversum.
Background and objective
Antibiotic stewardship programs have been implemented to optimize antibiotic prescribing which includes promoting the shortest effective duration of antibiotic treatment. Excessive use of antibiotics is associated with an increased risk of adverse events, Clostridioides difficile infections, multidrug-resistant pathogens, and unnecessary healthcare costs. Studies performed in mainly Anglo-Saxon countries showed that a substantial proportion of antibiotic treatments initiated in the hospital currently exceeds recommended durations. Approximately half of antibiotic exposure occurred after patients have been discharged, which emphasizes the importance of including post-discharge prescriptions when assessing the total duration of antibiotic therapy. Reliable data on antibiotic therapy is essential for establishing targets in the Netherlands to enhance antibiotic stewardship programs. Therefore, the objective of this study is to determine the total duration of antibiotic treatment for common infections and assess adherence to antibiotic treatment duration guidelines.
Methods
A retrospective cohort study was conducted among adult patients treated for urinary tract infections (UTI), respiratory tract infections (RTI), or skin and soft tissue infections (SSTI) in Tergooi Medical Centre. Data was extracted from the EHR from January 1, 2020 to December 31, 2022 using CTcue. Patients were included if they were admitted to any general medical ward for at least twelve hours and received antibiotics within the first 24 hours of admission. Antibiotic prescriptions, including those prescribed post-discharge, were linked to infectious diagnoses to calculate the length of therapy (LOT). Subsequently, the calculated LOT was compared against guidelines to assess excessive antibiotic duration.
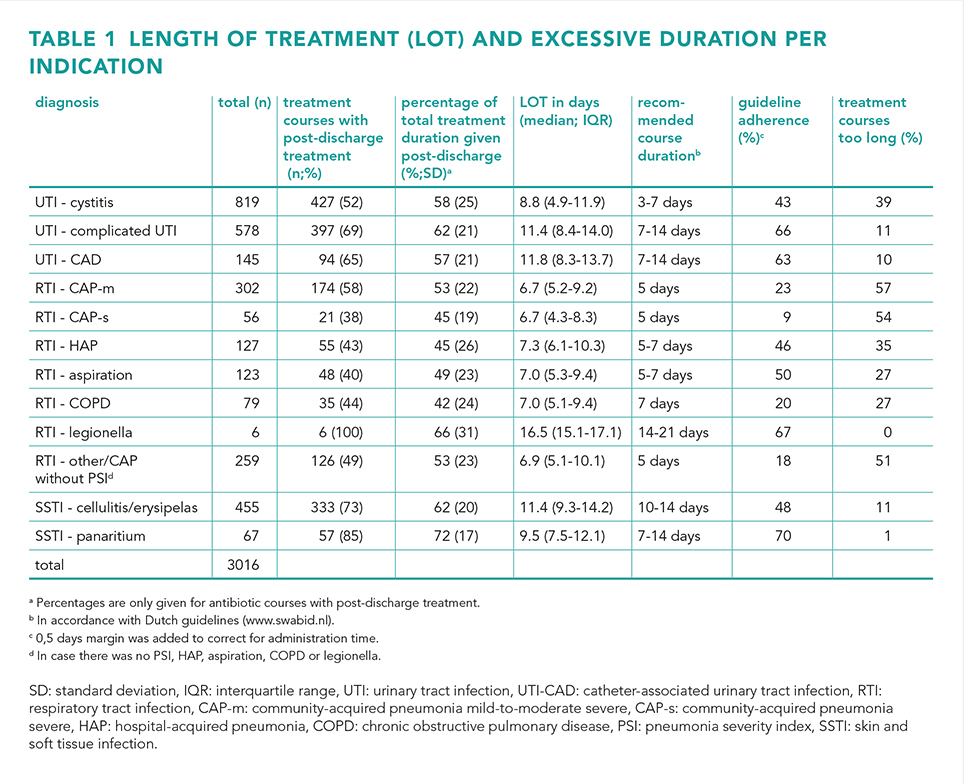
Results
A total of 3016 admissions were included, with 51% diagnosed with an UTI, 32% with an RTI and 17% with a SSTI. Guideline adherence regarding the total duration of antibiotic therapy varied from 9 to 70%, while the percentage of excessive treatment duration ranged from 0 to 57%. For UTIs, episodes of cystitis had the highest percentage of treatment courses that exceeded the recommended duration (39%). Overall, RTIs had the highest proportion of prescriptions that exceeding the guidelines, notably in mild-to-moderate-severe community-acquired pneumonia (57%). Additionally, 38-100% of the antibiotic courses were continued after discharge, which made out 42-72% of the total LOT.
Conclusion
While there was a considerable variability in guideline adherence, the majority of antibiotic therapies were found to be prescribed for too long. Furthermore, a substantial portion of antibiotics were prescribed post-discharge. Together, these findings highlight the added value of taking post-discharge antibiotics into account in antibiotic stewardship programs.
Dose optimization of vancomycin in paediatric post cardiac surgery patients: a population pharmacokinetic modeling study
J. Kamp a, D.J.E. Wannet b, P.P. Roeleveld c and D.J.A.R. Moes a
a Department of Clinical Pharmacy & Toxicology, Leiden University Medical Center.
b Department of Clinical Pharmacy, Meander Medical Center, Amersfoort,
c Department of Pediatric Intensive Care, Leiden University Medical Center.
Background
Vancomycin is a glycopeptide antibiotic used for the treatment of severe gram-positive infections. Despite decades of clinical experience, optimized dosing for vancomycin in paediatric populations still warrants further investigation. Children admitted to the paediatric intensive care unit (PICU) after cardiac surgery are often treated with vancomycin in case of infection. However, vancomycin dosing in this population is often challenging due to fluctuations in volume status, the use of diuretics or the use of extracorporeal membrane oxygenation (ECMO). The main aim of this study was therefore to describe vancomycin pharmacokinetics (PK) in paediatric cardiac surgery patients, and to optimize vancomycin dosing for this specific population. To this end, we performed a population pharmacokinetic modelling study, based on clinical data obtained from our paediatric cardiac surgery population.
Methods
A retrospective cohort study was performed with patients admitted to the PICU of the Leiden University Medical Center. Clinical data from post cardiac surgery PICU patients, receiving intravenous vancomycin between January 2020 and January 2021 were included in the analysis. Patients received vancomycin 10 mg/kg q.i.d., after which a trough concentration was sampled generally after the third dose. Pharmacokinetic data were used to develop a population PK model by using a non-linear PK modeling approach (NONMEM). In addition, potential covariates such as renal function, body weight and post menstrual age were tested on the model. The final model was subsequently used for vancomycin dose optimization.
Results
Thirty-five paediatric post cardiac surgery patients, contributing a total of 197 vancomycin blood samples were included in the dataset. A two-compartmental population pharmacokinetic model best described the data. In addition, renal function and body weight showed to significantly influence vancomycin pharmacokinetics. Model disposition parameters were: elimination clearance: 3.4 L/min at 70 kg, intercompartmental clearance: 0.575 L/min at 70 kg, central volume of distribution: 56.2 L/70 kg and peripheral volume of distribution: 248 L/70 kg (Fixed). Dose evaluations suggested non-linear dosing, with relatively lower per kg dosing for higher body weight patients to be optimal for our population.
Conclusion
We successfully developed a population pharmacokinetic model for vancomycin in post cardiac-surgery children. Vancomycin pharmacokinetics showed to be significantly influenced by serum creatinine and body weight. Furthermore, we suggest a new vancomycin dosing regimen based on an allometric scaling.
Clinical pharmacokinetics and practical dosing recommendations of vancomycin in patients with neutropenia: a systematic review
Madina Gadaborcheva a, Ron J. Keizer b, Heidi S.M. Ammerlaan c, Sophie L. Stocker def and Maarten J. Deenen ag
a Department of Clinical Pharmacy, Catharina Hospital, Eindhoven.
b Insight RX, San Francisco, CA 94104, USA.
c Department of Internal Medicine, Catharina Hospital, Eindhoven.
d School of Pharmacy, Faculty of Medicine and Health, The University of Sydney, Australia.
e Sydney Institute for Infectious Diseases, The University of Sydney, NSW, Australia.
f Department of Clinical Pharmacology & Toxicology, St Vincent's Hospital Sydney, Australia.
g Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
Background
Potentially serious infections are common in patients with neutropenia. Especially neutropenic patients are vulnerable, therefore adequate pharmacotherapeutic target attainment of these infections is crucial. Vancomycin is extensively used for the treatment of gram-positive infections in patients with neutropenia. However, subtherapeutic vancomycin concentrations are often observed in neutropenic patients following standard dosing. We aimed to systematically review the current evidence on the pharmacokinetics of vancomycin in patients with neutropenia and to translate the available evidence into practical dosing recommendations to improve vancomycin dosing for neutropenic patients.
Methods
A systematic literature search was performed in PubMed and Embase from database inception to February 1st, 2023. English studies reporting on pharmacokinetic data of vancomycin in neutropenic patients (even if a subset of the total study population) were included. Studies were excluded if (i) insufficient pharmacokinetic or clinical data were reported; (ii) they did not specify if patients were neutropenic; or (iii) were review articles, editorials or only published as abstracts. All available pharmacokinetic parameters were retrieved, and whenever possible, vancomycin clearance (CL) and volume of distribution (Vd) was assessed for both patients with and without neutropenia. In addition, vancomycin dosing recommendations for neutropenic patients were derived.
Results
Twelve articles were identified in which 6/12 (50%) were performed only in patients with neutropenia, and 50% in both patients with and without neutropenia. A consistent observation across all studies was that patients with neutropenia often had subtherapeutic vancomycin concentrations following standard dosing regimens. This was due to a median (range) 28% (23-34%) increased clearance of vancomycin in patients with neutropenia (3.8 L/h [3.6-4.0 L/h]) versus patients without neutropenia (3.0 L/h [2.8-3.1 L/h]). Neutropenia was an independent risk factor for increased clearance of vancomycin. The Vd ranged from 42-122 L and was generally similar in patients with and without neutropenia. Based on the effect of neutropenia on vancomycin clearance, an initial pragmatically vancomycin dose increase of approximately 30%, followed by TDM and further individual dose titration is suggested to improve attainment of vancomycin therapeutic targets.
Conclusion
Standard vancomycin dosing regimens generally result in subtherapeutic drug exposure in patients with neutropenic because these patients have increased clearance of vancomycin. To achieve adequate systemic exposure, the vancomycin dose should be increased by 30% in patients with neutropenia, followed by therapeutic drug monitoring to inform subsequent dosing decisions. Since the volume of distribution is not affected by neutropenia no change in the initial loading dose is required.
Clinical validation of a dried blood spot method for simultaneous measurement of vancomycin and creatinine
M. Hassanzai a, S. Bahmany a, H.A.W. van Onzenoort b, B.C.P. Koch a and B.C.M. de Winter a
a Department of Hospital Pharmacy, Erasmus MC University Medical Center, Rotterdam.
b Department of Pharmacy, Radboud University Medical Centre, Radboud Institute for Health Sciences, Nijmegen.
Background
Vancomycin is widely used in Outpatient Parenteral Antimicrobial Therapy (OPAT) services, however a drawback of its use is the need for therapeutic drug monitoring. Traditional blood sampling consists of drawing blood trough a venepuncture by a nurse, often performed at the hospital or at an phlebotomy facility. An alternative method, dried blood spot (DBS) sampling, involves collecting a small drop of capillary blood by a finger prick. This technique is less invasive and allows for self-sampling at home.
Objective
The objective of this study is to clinically validate a DBS method for the measurement of vancomycin and creatinine in comparison to the venepuncture method.
Methods
Hospitalized adults treated with intravenous continuous or intermittent vancomycin were eligible to participate. Blood sampling consisted of one venepuncture and one finger prick (DBSfinger). Preferably, the time between the finger prick and the venepuncture was as short as possible. Through levels were obtained for intermittent dosing. The DBS whole-blood from the capillaries was spotted onto the Whatman 903 filtrate card paper and dried in a desiccator. DBS samples analyses were performed using the LC-MS/MS method which was previously validated based on FDA and EMA guidelines. The venepuncture samples were used to measure vancomycin and creatinine concentrations in plasma via enzymatic immunoassay. In addition, patients also were asked to state their preference for one of the two sampling methods.
Results
The study involved a final analysis of 39 patients for the clinical validation. The difference between plasma and DBSfinger was ≤ 20% for 76% of the vancomycin samples. The difference between plasma and DBSfinger was ≤ 20% for 90% of the creatinine samples. No effect was observed from haematocrit, the DBS filter paper and the use of anti-coagulant agents. There was no need for the use of a correction factor, as the data fulfil the criteria from the IATDMCT guidelines. The time difference between DBS and venepuncture sampling varied between −130 to 214 minutes, explaining the lower agreement between DBS and venepuncture concentrations of the vancomycin samples. Most patients asked (18 out of 31) preferred a finger prick over a venepuncture, one patient preferred a venepuncture over a finger prick and 12 patients indicated no preference.
Conclusion
This is the first study that successfully clinically validated a DBS sampling method for simultaneous measurement of vancomycin and creatinine for use in clinical practice.
Best practices, implementation and challenges of outpatient parenteral antimicrobial therapy: results of a worldwide survey among healthcare providers
M. Hassanzai a, F. Adanç a, B.C.P. Koch ab, N.J. Verkaik c, J. van Oldenrijk d, J.L. de Bruin e, B.C.M. de Winter ab and H.A.W. van Onzenoort f
a Department of Hospital Pharmacy, Erasmus MC University Medical Center, Rotterdam.
b CATOR: Center for Antimicrobial Treatment Optimization Rotterdam.
c Department of Medical Microbiology and Infectious Diseases, , Erasmus MC University Medical Center, Rotterdam.
d Department of Orthopaedics and Sports Medicine, Erasmus MC University Medical Center, Rotterdam.
e Department of Vascular Surgery, Erasmus University Medical Center, Rotterdam.
f Department of Pharmacy, Radboud University Medical Centre, Radboud Institute for Health Sciences, Nijmegen and Department of Clinical Pharmacy and Toxicology, Maastricht University Medical Center+.
Background
Outpatient Parenteral Antimicrobial Therapy (OPAT) is considered a patient-friendly and cost-effective practice and is nowadays part of regular care in many countries. Patients receiving antimicrobial therapy in the outpatient setting can however be still at risk for developing adverse events. Due to extensive variations in practice, guidelines have been developed to minimize the risks associated with OPAT. In this first worldwide survey, we explored the current OPAT services around the world, adherence to OPAT recommendations and identified best practices and challenges from different perspectives.;
Methods
An online survey (LimeSurvey) was conducted from April to September 2022. The survey was distributed by addressing (inter)national professional associations and organizations with OPAT as one of their areas of interest. The survey consisted of 34 questions including demographics, characteristics of the OPAT service, role of pharmacy, future developments, and respondents' views on improvements as well as best practices.
Results
A total of 126 responses from 103 healthcare facilities and 28 countries were included. The majority of the respondents were from Europe (51%), followed by Asia (18%) and Oceania (15%). 60% of the respondents were pharmacists. 78% of the respondents stated that their facility provides antimicrobial therapy in the outpatient setting, whereas 22% did not. The main reasons for not having an OPAT service were a lack of financial resources (25%) and time (18%). 42% of the hospitals with OPAT services had a specialized OPAT service with a designated team, while 14% lacked specialized services and 22% had a partially specialized team in place. In facilities with a specialized OPAT service with a designated OPAT team the number of mandatory ID consultation before discharge was higher compared to overall response. In addition, clinical monitoring by an ID specialist or OPAT team member, the frequency of monitoring and availability of an OPAT registry was also higher. A multidisciplinary team's presence and the use of elastomeric pumps were commonly noted as best practices. On the other hand, respondents experienced difficulties with reimbursement and lack of standardization in the screening, follow-up and monitoring of patients.
Conclusion
This survey provides a better understanding of the implementation and practices of OPAT services globally and describes best practices and the challenges from different professionals involved in OPAT.
Antithrombotic stewardship in Dutch hospital pharmacies: a questionnaire based survey
J. Graafsma a, J.E. Klopotowska bc, H.J. Derijks d, E.M.W. van de Garde ef, H.L. Hoge gh, F. Karapinar-Carkit i and P.M.L.A. van den Bemt a
a Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen.
b Department of Medical Informatics Amsterdam UMC, University of Amsterdam.
c Amsterdam Public Health Institute.
d Department of Pharmacy, Jeroen Bosch Hospital, Den Bosch.
e Department of Pharmacy, St. Antonius Hospital, Utrecht/Nieuwegein.
f Division Pharmacoepidemiology and Clinical Pharmacology, Utrecht University.
g Department of Pharmacy, Wilhelmina Hospital, Assen.
h Gaston Medical, Eindhoven.
i Department of Clinical Pharmacy & Toxicology, Maastricht University Medical Center+.
Background
Antithrombotics require careful monitoring to prevent unnecessary complications. The national Dutch guideline ‘Landelijke Standaard Ketenzorg Antistolling’ (LSKA) published in 2012, provides guidance on how to ensure safe use of antithrombotics via the so-called antithrombotic stewardship. Antithrombotic stewardship includes the following tasks: professional’s education, consultation on complex patients, medication reviews, drafting and maintenance of protocols, patient counseling and care transition optimization. At present, it is unknown to what extent antithrombotic stewardship is implemented in Dutch hospitals and how clinical decision support systems (CDSSs) are used to support the aforementioned tasks.
Methods
A multicentre, cross-sectional study using a semi-structured questionnaire-based survey was sent out to 12 hospital pharmacists from different hospital types and regions in the Netherlands. The primary outcome was the degree of antithrombotic stewardship adoption, expressed as the number of tasks implemented per hospital and the degree of implementation per task. Secondary outcomes were the characteristics of CDSSs used in the hospitals for monitoring of antithrombotics. Descriptive statistics were used to analyse the data.
Results
The survey was completed by ten hospital pharmacists (response rate 83%), representing one university hospital, six teaching hospitals and three general hospitals. All hospitals adopted antithrombotic stewardship to a certain degree, but none reached full adoption. Most hospitals (n = 4) adopted three of the six mentioned tasks. The tasks with the highest uptake were: drafting and maintenance of protocols (n = 10), professional’s education (n = 6) and consultation on complex patients (n = 6). The tasks with the lowest uptake were care transition optimization (n = 2), medication reviews (n = 1) and patient counselling (n = 1).
Regarding the use of CDSSs, all ten hospitals used clinical rules in addition to basic medication surveillance based on ‘G-Standaard’ alerts. The most frequently employed clinical rules were: identification of patients with impaired renal function using a low molecular weight heparin (LMWH) or a direct oral anticoagulant (DOAC) (n = 4) and detecting patients using a vitamin K antagonist (VKA) together with a LMWH having an International Normalized Ratio (INR) within therapeutic range (n = 3).
Conclusion
Findings of this study reveal that, more than 10 years after publication of the LSKA, the tasks advised by the LSKA are just partly adopted by antithrombotic stewardships. Furthermore, the tasks implemented vary across the hospitals. Only a few clinical rules are used and these reflect basic CDSS. To optimize safe use of antithrombotics, antithrombotic stewardship needs to be adopted completely and broader use of more advanced CDSS may facilitate this.
Pharmacological effects of CFTR modulation in CF patients after lung transplantation: interim results of the multicentre KOALA study
Carina Hansen a, Klara Visser b, Anna van Gemert b, Bart Luijk c, Merel Helemons d, Hester van de Vaart b, Harry Heijerman c and Erik Verschuuren b
a Department of Clinical Pharmacy & Pharmacology, University Medical Center Groningen.
b Department of Respiratory Diseases, Tuberculosis and Lung Transplantation, University of Groningen, University Medical Center Groningen.
c Department of of Respiratory Diseases, University of Utrecht, University Medical Center Utrecht.
d Department of Respiratory Diseases, Erasmus MC Transplant Institute, Erasmus University, University Medical Center, Rotterdam.
Background and objective
Cystic fibrosis leads to dysfunction of multiple organ systems, lung transplant (LTx) patients experience extrapulmonary symptoms even after lung transplantation, and might benefit from cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy. The multicentre KOALA study aims to assess the extrapulmonary effect of CFTR modulators in LTx patients with CF. Here, we report the short-term interim results of a single site.
Methods
Multicentre observational study in the Netherlands. Patients with at least one F508del mutation, who underwent a single, bilateral, lung-liver or heart-lung transplantation in the University Medical Center Groningen (UMCG), Erasmus Medical Center or University Medical Center Utrecht were provided CFTR modulator therapy upon written informed consent. Outcomes included change in dose of the calcineurin inhibitor (tacrolimus or cyclosporin A) and/or mTOR inhibitor (everolimus/sirolimus) as well as adverse events. Long-term outcomes (to be reported later) will include change in BMI, HbA1c, SNOT-22 score, abdominal complaints score, lung function 2, 4, 8, 12 weeks and 1 year after the start of CFTR modulator therapy.
Results
At t = 10 weeks, 10 patients (mean age 39.5, 80% male, starting Kaftrio on average 12.9 years after LTx) were included. Tacrolimus dose was highly variable. Median (IQR) tacrolimus level at baseline was 7 (2.8) ug/L and mean increase at 2 weeks was 0.8 ug/L. The majority of patiënts had adverse events in the first 2 weeks after initiation: headache (63%), gastro-intestinal complaints (63%), dysregulated glucose (25%). Three patients experienced severe itching and erythema (all solved by antihistamines). Two patients experienced severe muscle aches and severe headache and one had a decreased appetite resulting in discontinuing at 5 weeks despite dose reduction at 4 weeks.
Conclusion
CFTR modulators in LTx patients with CF have minimum impact on calcineurin inhibitor level, most side effects were reversible within 2 weeks yet itch and muscle pain were notable.
Implications of tioguanine dosing in IBD patients with a TPMT deficiency
Debbie S. Deben a, Luc J.J. Derijks b, Bianca J.C. van den Bosch c, Rob H. Creemers de, Annick van Nunen d, Adriaan A. van Bodegraven de and Dennis R. Wong a
a Zuyderland Medical Centre, Department of Clinical Pharmacy, Clinical pharmacology and Toxicology, Sittard-Geleen-Heerlen.
b Máxima Medical Centre, Department of Clinical Pharmacy and Pharmacology, Veldhoven.
c Maastricht University Medical Centre (MUMC+), Department of Clinical Genetics.
d Zuyderland Medical Centre, Department of Gastroenterology, Sittard-Geleen-Heerlen.
e Maastricht University Medical Centre (MUMC+), Department of Gastro-enterology.
Background and objective
Tioguanine is metabolised by less enzymatic steps compared to azathioprine and mercaptopurine, without generating 6-methylmercaptopurine ribonucleotides. However, thiopurine S-methyl transferase (TPMT) plays a role in early toxicity in all thiopurines. We aimed to describe the hazards and opportunities of tioguanine use in inflammatory bowel disease (IBD) patients with aberrant TPMT metabolism and propose preventative measures to safely prescribe tioguanine in these patients.
Methods
In this retrospective cohort study, all determined TPMT genotypes (2016-2021) were evaluated for aberrant metabolism (i.e. intermediate and poor TPMT metabolisers). Subsequently, all IBD patients on tioguanine with aberrant TPMT genotypes were evaluated for tioguanine dosages, adverse drug events, lab abnormalities, treatment duration and effectiveness.
Results
TPMT genotypes were determined in 485 patients of whom 50 (10.3%) and 4 patients (0.8%) were intermediate and poor metabolisers, respectively. Of these patients, 12 intermediate and 4 poor TPMT metabolisers had been prescribed tioguanine in varying doses. In one poor TPMT metaboliser, tioguanine 10 mg/day induced delayed pancytopenia.
In general, reduced tioguanine dosages of 5 mg/day for intermediate TPMT metabolisers, and 10 mg two-weekly for poor TPMT metabolisers, resulted in a safe, long-term treatment strategy.
Conclusion
Diminished or absent TPMT enzyme activity was related with a pharmacokinetic shift of tioguanine metabolism which is associated with relatively late occurring myelotoxicity in patients on standard tioguanine dose. However, in strongly reduced dose regimens with strict therapeutic drug and safety monitoring, tioguanine treatment remained a safe and effective option in IBD patients with dysfunctional TPMT.
Pharmacokinetic data of atazanavir/ritonavir in second-line treatment of children living with HIV: results from the CHAPAS4-trial
Anne Kamphuis a, Hylke Waalewijn ab, Alexander Szucbert c, Chishala Chabala d, Mutsa Bwakura-Dangarembizi e, Shafic Makumbi f, Joan Nangiya g, Vivian Mumbiro e, Veronica Mulenga d, Victor Mussiime g, David Burger a, Diana Gibb c, Angela Colbers a and the CHAPAS4 trial team
a Department of Pharmacy, Radboud Research institute for Medical Innovation (RIMI), Radboudumc, Nijmegen.
b Division of Clinical Pharmacology, Department of Medicine, University of Cape Town, Cape Town, South Africa.
c Medical Research Council Clinical Trials Unit at University College London, London, United Kingdom.
d University Teaching Hospital, Lusaka, Zambia.
e University of Zimbabwe Clinical Research Centre, Harare, Zimbabwe.
f Joint Clinical Research Centre, Mbarara, Uganda.
g Joint Clinical Research Centre, Kampala, Uganda.
Background and objective
In 2022, 1.5 million children were living with HIV worldwide. As children need life-long antiretroviral therapy, the question is raised whether the current second-line antiretroviral regimens are optimal with regard to maximizing children’s health gains. Atazanavir/ritonavir (ATV/r) is recommended by the World Health Organization for children living with HIV as a preferred boosted protease inhibitor for second-line treatment. ATV/r has been evaluated in African children enrolled in the randomized controlled CHAPAS4 trial (#ISRCTN22964075), where second-line treatment options for children with HIV were investigated. We did a pharmacokinetic (PK) sub-study within CHAPAS4 to evaluate the ATV/r exposure in children with HIV.
Methods
Children living with HIV aged 3-15 years failing first-line antiretroviral therapy were enrolled. In this sub-study children in weight bands 14-19.9 and 20-24.9 kg received 200/75 mg ATV/r and children 25-34.9 and ≥ 35 kg received 300/100 mg ATV/r. At steady-state, 8 ATV/r plasma PK samples were taken over 24 hours after observed ATV/r intake. The primary pharmacokinetic parameters were the area under the concentration-time curve over 24 hours (AUC0-24h), maximum concentration (Cmax), and the concentration 24 hours after intake (Ctrough), calculated using non-compartmental analysis. Reference adult PK data were used for comparison. The individual target trough concentration (Ctrough) was defined as 0.15 mg/L (EC90). Statistical analysis was performed using ANOVA on log-transformed values to check for differences in ATV/r AUC0-24h and Ctrough in the different weight bands and nucleoside reverse transcriptase inhibitor (NRTI) backbones.
Results
A total of 61 children were included in this sub-study. Seven children were excluded due to non-adherence, RTV dose deviation or dosing at the wrong time, leaving 54 eligible PK profiles to evaluate. For the whole study population, the Geometric Mean (GM), (CV%) AUC0-24h was 44.3 mg*h/L (47%) and GM (CV%) Cmax was 4.6 mg/L (47%), which is comparable to the adult reference values. The AUC0-24h was significantly higher in children weighing 25-34.9 kg (61.1 h*mg/L) compared to children in the 14-19.9 kg and 20-24.9 kg weight bands (34.1 and 33.2 h*mg/L, respectively); P-values < 0.05. There was no difference in AUC0-24h when comparing the different backbones. The GM (CV%) Ctrough was 0.48 mg/L (70%), which is below the adult reference Cmin (0.64 mg/L), but the Ctrough target of 0.15 mg/L was achieved in all subjects.
Conclusion
This PK sub-study shows that the exposure of ATV/r taken with food in children 3-15 years weighing ≥ 14 kg on second-line treatment is comparable to adult reference data.
Design and mechanics of five major human milk drug research biobanks: time to cross borders and barriers
P. Mian ab, K.M. Watt c, C. Chambers d, K. Allegaert efg, K. Krutsch h, S. Ito i and A.N. Edginton a
a School of Pharmacy, University of Waterloo, Waterloo, Ontario, Canada.
b Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen and University of Groningen.
c Division of Clinical Pharmacology, Department of Pediatrics, Spencer Fox Eccles School of Medicine, University of Utah, Salt Lake City, Utah, USA.
d Department of Pediatrics, University of California, San Diego, La Jolla, California, USA.
e Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
f Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, Leuven, Belgium.
g Department of Hospital Pharmacy, Erasmus MC, Rotterdam.
h Department of Obstetrics and Gynecology, Texas Tech University Health Sciences Center, Amarillo, Texas, USA.
i Division of Clinical Pharmacology and Toxicology, Hospital for Sick Children, Toronto, Ontario, Canada.
Background and objective
To better understand and inform complex decisions about maternal medication during lactation several human milk drug research biobanks have been established worldwide. The aim of this study was to describe the biobanks’ strategies to overcome the barriers they have each identified to drive improvements in milk quality, safety, and clinical use.
Methods
A literature search was performed on PubMed on April 3rd to identify human milk drug research biobanks. A search was performed, with a combination of different terms like: ‘Milk, Human’ and ‘Milk, Banks’. Studies were labelled as relevant when information on drug milk concentrations within a biobank was available. In addition, the principal investigators of the included biobanks were asked whether they had any knowledge on possible human milk drug biobanks not obtained with the literature search. Principal investigators were interviewed on logistical (ethics, resources, equipment), methodological (e.g., recruitment, milk sampling strategy), and supporting information (e.g. maternal/infant health records, additional matrices).
Results
Five human milk drug research biobanks were identified and the principal investigators were interviewed. Four biobanks were located in North America and one in Europe. All, but one, did not pre-define the medication to be investigated, but rather collected samples broadly and allowed the subsequent analyses to depend on the specific exposures of interest. Only one biobank developed study-defined sampling schedules depending on the drug characteristics, while the other biobanks performed opportunistic sampling. However, it must be noted that for the biobank with sampling schedules, opportunistic samples are accepted if participants are unable/unwilling to follow the schedule. Two biobanks collect purposeful samples, while three collect samples for general research purposes. Besides similarities between the biobanks, differences existed, for example, in where milk samples were collected (50% home versus 50% hospital collection), and the drug groups studied (non-medical substances, commonly used versus non-commonly used medications).
Conclusion
All human milk drug research biobanks are well-characterized, accessible research sources that can contribute to a better understanding in the potential effects of maternal medication and on the health and development of the breastfed infants.
ICU admissions due to olanzapine post-injection delirium/sedation syndrome: a case series and review of the literature
Sietske A. Kochen a, Charlotte S. Hakkers b, Freek van Gorp a, Dylan W. de Lange b and Lenneke E.M. Haas a
a Diakonessenhuis, Utrecht.
b UMC Utrecht.
Background and objective
Olanzapine long-acting injection (OLAI) is a commonly used antipsychotic administration in the treatment of schizophrenia. Post-injection delirium/sedation syndrome (PDSS) is a potential side effect of this intramuscular depot. This report presents a case series of ten patients admitted to the ICU due to PDSS and provides an overview of the current literature outlining the key aspects of managing this syndrome in a critical care setting.
Methods
All patients admitted to the ICU of a Dutch teaching hospital with PDSS after OLAI, between March 2017 and July 2021, were retrospectively included.
Results
Ten patients with PDSS were included. In eight patients, the onset of symptoms occurred within 1 hour after administration. All ten patients had altered consciousness (consisting of sedation or agitation or a combination of both) during admission and all patients showed tachycardia but were hemodynamically stable. Other symptoms that were seen included extrapyramidal symptoms, ataxia, dysarthria, hypo- or hypertension and QTc prolongation. All patients received conservative treatment and could be discharged within 48 hours.
Conclusion
PDSS is a potential side effect of OLAI. Symptoms, including altered consciousness and tachycardia, occur soon after administration. All patients received conservative treatment and were discharged within 48 hours.
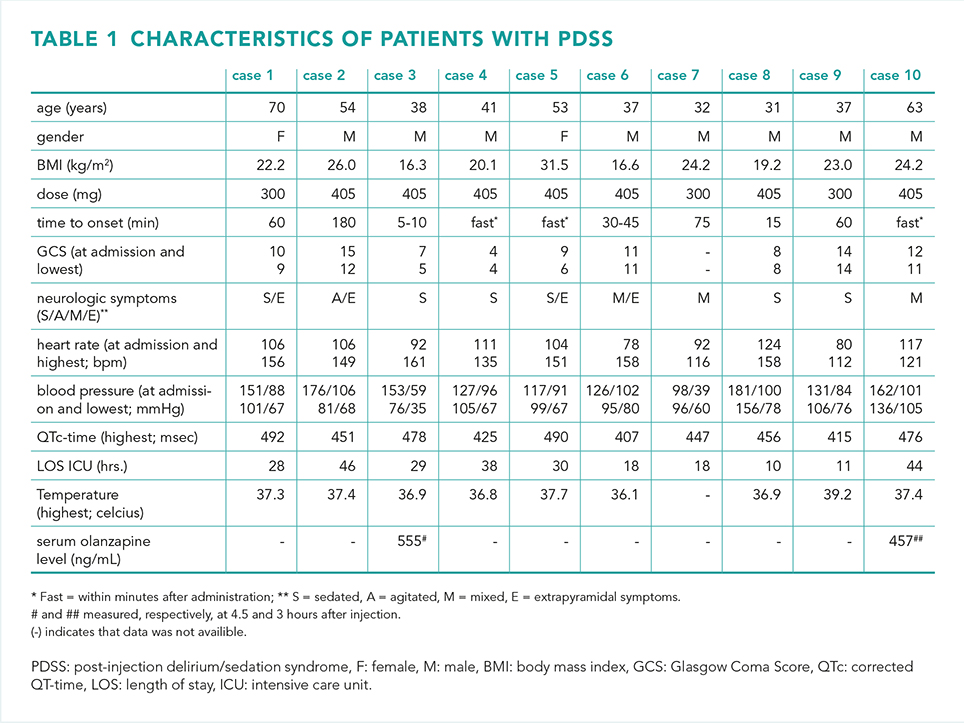
Performance of multiple trigger tools in identifying medication-related hospital readmissions
Amit Singh a†, Nikki Lips b†, Daniala Weir c and Fatma Karapinar-Carkit ad
a Department of Clinical Pharmacy, OLVG Hospital, Amsterdam.
b Department of Internal Medicine, OLVG Hospital, Amsterdam.
c Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
d Department of Clinical Pharmacy and Toxicology, MUMC+ Hospital, Maastricht.
† These authors contributed equally to this work and share the first authorship.
Background and objective
The Dutch polypharmacy guideline recommends using a trigger tool to identify medication-related hospital (re)admissions. Many trigger tools exist for this purpose, especially in older patients. Yet, the effectiveness of these trigger tools and clinical applicability remains uncertain. This study evaluates these tools' performance in identifying medication-related readmissions (MRRs).
Method
In a single-centre cross-sectional study, we analysed data from a previous study assessing readmissions within 30 days after discharge across seven clinical departments. In this prior study, a panel of physicians and pharmacists retrospectively assessed readmissions as medication-related, including preventability. This current study employed four trigger tools (Screening Tool to Alert doctors to Right Treatment / Screening Tool of Older Person’s Presciptions [START-STOPP] criteria, OPtimising thERapy to prevent Avoidable hospital readmissions in Multimorbid older people
[OPERAM], Adverse Drug Reaction [ADR]-tool, and QUick Assessment of Drug-Related Admissions over Time [QUADRAT]) on clinically adjudicated MRRs. The START-STOPP criteria focus on under- and overtreatment, OPERAM on multiple causes of medication related (re)admissions, while ADR and QUADRAT tools focus on side effects. The tools include explicit (e.g., diuretics and dehydration) and implicit general triggers (e.g., addressing double medication).
A hospital pharmacist, internal medicine resident, and pharmacy student applied these trigger tools to clinically adjudicated MRRs in duplicate. The primary outcome was each tool's performance in identifying MRRs with triggers. Secondary outcomes included assessing the performances of these tools in identifying MRRs based on recognition of MRRs by attending physicians, potential preventability of the MRRs and age of patients. Descriptive data-analysis was used.
Results
There were 181 MRRs of which 159 (88%) were recognized as medication-related by the attending physician at the time of readmission. Among the 181 MRRs, the OPERAM trigger tool identified 92% of MRRs (62% explicit and 30% implicit triggers). The ADR and QUADRAT tools identified 51% and 76% of MRRs, respectively. START-STOPP criteria identified the fewest MRRs (7%). The tools were more effective in identifying MRRs recognized by attending physicians, with greater identification of non-preventable MRRs, except for START-STOPP criteria. No differences were observed in identified MRRs between patients below and above 70 years.
Conclusion
Multiple trigger tools were applied to real-life patient data. START-STOPP criteria, ADR-tool (recommended by the Dutch polypharmacy guideline) and QUADRAT were unsuccessful in identifying MRRs in this study. OPERAM performed the best but included many implicit triggers necessitating substantial reviewer knowledge to assess medication-relatedness of readmissions. Consequently, even OPERAM is unsuitable as a quick screening tool for identifying MRRs in this study.
The effect of digital clinical decision support on pharmacotherapy in hospitalized (morbidly) obese patients: a prospective intervention study
A .Keyany a, I. Groenen b and B. Maat a
a Department of Hospital Pharmacy, Elisabeth TweeSteden Hospital (ETZ), Tilburg.
b Department of Pharmaco-epidemiology and pharmacotherapy, Utrecht University.
Background
The pharmacokinetics and -dynamics of medication can be altered in (morbidly) obese patients. Standard medication doses in patients with (morbid) obesity may be suboptimal and adjustments based on body mass index (BMI) or body weight (BW) may be needed. Digital clinical decision support (eCDS) may help optimize pharmacotherapy in these patients.
Objective
The aim of this study was to assess the effect of eCDS on adjustments in pharmacotherapy based on BMI or BW in hospitalized (morbidly) obese patients.
Methods
This prospective intervention study with retrospective baseline measurement took place in hospitalized ETZ patients ≥ 18 years with BMI ≥ 30 kg/m² and/or BW ≥ 90 kg from 1/1/2022-30/9/2022 (pre-eCDS group) and from 10/10/2022-25/11/2022 (post-eCDS group). In the intervention period the hospital pharmacy recommended pharmacotherapy adjustments to prescribers based on eCDS. eCDS is a tool, integrated in the electronic hospital record (Epic), that detected patients whose medication order(s) potentially needed to be adjusted to BMI or BW. Study outcomes were (i) number of post-eCDS patients with ≥ 1 medication orders resulting in a recommendation for adjustment, including medication details, (ii) number and percentage of recommendations that actually led to an adjustment in pharmacotherapy, including reasons for rejecting a recommendation and (iii) number percentage of patients whose pharmacotherapy was adjusted to BMI or BW pre-eCDS versus post-eCDS.
Results
Post-eCDS 328 patients were detected with ≥ 1 medication orders resulting in a recommendation for adjustment. In the intervention period 186 of 349 (53.3%) recommendations actually led to an adjustment in pharmacotherapy. The majority of eCDS triggers, recommendations and adjustments were for nadroparin, respectively 95% (796/842), 93% (324/350) and 89% (163/186). The main reason for not accepting a recommendation by a physician was discharge from hospital: 90.8% (148/163). In the post-eCDS group pharmacotherapy was significantly more often adjusted to BMI or BW: 77.7% (912 of 1,173 medication orders) post-eCDS versus 58.2% (3,519 of 6,049 medication orders) pre-eCDS (P < 0.0001).
Conclusion
Implementation of digital CDS in hospital pharmacy led to a significant increase in medication orders adjusted to BMI or BW, in (morbidly) obese patients.
Inpatients’ medicine information needs: an analysis of semi-structured interviews with patients and members of the patient council
S. Wilkes , A.S.E van der Sman, I.H. van der Sijs, H.P.M. van der Kuy and R. J. Zaal
Erasmus MC, University Medical Center Rotterdam, Department of Hospital Pharmacy.
Background and objective
Patients often receive new medication or experience medication alterations during hospital admission. In order to achieve optimal pharmaceutical treatment and patient satisfaction, it is important that patients are informed about their pharmacotherapy. Current studies about this topic predominantly included patients admitted to a psychiatry, cardiology or internal ward. Currently, little is known about medicine information needs for patients during hospital admission in a general inpatient population. Therefore, the aim of this study is to explore the medicine information needs for hospitalized patients. The secondary aim is to study the optimal method for providing medicine information: the best moment for providing medication information, by whom and how.
Methods
A qualitative study using semi-structured interviews with patients who were admitted to a tertiary hospital and members of the patient council. Purposive sampling was used, and participants were included until data saturation was reached.
Results
Interviews were conducted with ten patients and two members of the patient’s council. Patients wanted to be informed about the name of the medication, the reason for starting it and how to use it. Patients also mentioned side effects as important information, but not all patients want to receive this information. Medicine information was preferred before the first administration. Patients differed in their preferences for the healthcare professional providing the medicine information, for example the prescriber, pharmacy employees or the nurse. But they unanimously mentioned that the professional should have sufficient medicine knowledge. Preferably, the information should be given both face to face supported by written information, on paper or digitally accessible.
Conclusion
Health care professionals should proactively provide medicine information to hospitalized patients. It is necessary to implement this in regular practice, and support oral information about newly introduced medication with printed or digitally accessible information.
Patient-led medication reconciliation via the hospital patient portal
E.E. Hendriks, K.B. Gombert-Handoko
Leiden University Medical Center.
Background and objective
Medication reconciliation is a process which is commonly conducted by pharmacy technicians or physicians. This is an important, though time-consuming process. This study was conducted to explore whether the hospital’s electronic health portal is suitable for patients to perform medication reconciliation by themselves.
Methods
In this study, patients undergoing elective cardiological admissions at the LUMC were approached over a sixteen-week period from February 12 until May 31 2023. The main source for medication in the electronic health record was derived from the Landelijk Schakel Punt (LSP). Patients received an email referral to 'mijnlumc' a week before their hospital admission, where they were requested to verify their medication. The pharmacist updated the medication list and documented the patient's verification in the electronic health record. The primary endpoint was the number of patients who responded on the question to perform the medication reconciliation. Secondary endpoints were the number of medication discrepancies per patient and the time investment compared to regular care.
Results
A total of 647 patients were included, on average 43 patients per week. In total 52% (335/647) of patients verified their medication before admission. Among responsive patients, half (172/335) had accurate medication lists, while the other half (163/335) reported an average of two discrepancies each. Age and medication quantity did not significantly affect responsiveness. The average time investment per patient for the hospital pharmacy to facilitate the patient to do his own medication reconciliation was 4 minutes. When the patient responded this took another 2 minutes. The average time for a standard medication reconciliation is 16 minutes. So when a patient responded 10 minutes per patient can be saved. For our cardiology department with on average 43 elective admissions per week, and a 50% response 3,6 hours of health care professional time per week is saved by this method.
Conclusion
Independent medication verification offers several benefits, including increased patient autonomy and the flexibility for the patient to review medication at a convenient time. This study shows that 50% of elective cardiology patients were able to perform medication reconciliation by using the hospital patient portal. Furthermore, a considerable amount of hours of health care professional time could be saved by this method.
Factors influencing pharmacists' clinical decision-making in pharmacy practice
J.F.Mertens a, E.S. Koster b, V.H.M. Deneer bc, M.L. Bouvy b and T. van Gelder a
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
b Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences (UIPS), Department of Pharmaceutical Sciences, Utrecht University.
c Department of Clinical Pharmacy, Division of Laboratories, Pharmacy, and Biomedical Genetics, University Medical Center Utrecht.
Background
Pharmacists’ clinical decision-making is considered a core process of pharmaceutical care in pharmacy practice, but little is known about the factors influencing this process.
Objective
To identify factors influencing clinical decision-making among pharmacists working in pharmacy practice.
Methods
Semi-structured interviews were conducted with pharmacists working in primary, secondary, and tertiary care settings in the Netherlands between August and December 2021. A thematic analysis was conducted using an inductive approach. The emerged themes were categorized into the Capability-Opportunity-Motivation-Behaviour (COM-B) model domains.
Results
In total, 16 pharmacists working in primary care (n = 7), secondary care (n = 4) or tertiary care (n = 5) were interviewed. Factors influencing pharmacists’ capability to make clinical decisions are a broad theoretical knowledge base, clinical experience, and skills, including contextualizing data, clinical reasoning, and clinical judgment. The pharmacy setting, data availability, rules and regulations, intra- and interprofessional collaboration, education, patient perspectives, and time are mentioned as factors influencing their opportunity. Factors influencing pharmacists’ motivation are confidence, curiosity, critical thinking, and responsibility. Influencing factors are organized in the COM-B model (figure 1).
Conclusion
The reported factors covered all domains of the COM-B model, implying that clinical decision-making is influenced by a combination of pharmacists’ capability, opportunity, and motivation. Addressing these different factors in pharmacy practice and education may improve pharmacists’ clinical decision-making, thereby improving patient outcomes.
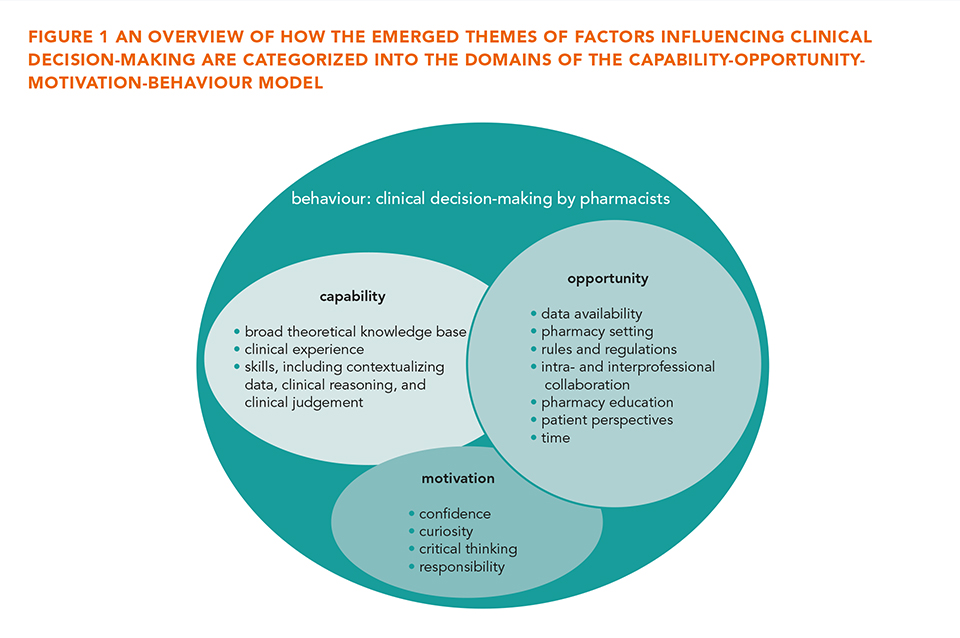
Daily vitamin supplementation in hemodialysis patients results in high levels of vitamin B1 and B6
F.J. van den Oever a, L. van Rooi a, E.C. Vasbinder a, M. Kerskes b, A. Aarnoudse b, M. Hengst c, H. van Hamersvelt d and J. van der Leeuw a
a Franciscus Gasthuis and Vlietland Hospital, Rotterdam.
b Catharina Hospital, Eindhoven.
c Elisabeth Tweesteden Hospital, Tilburg.
d Radboud University Medical Center, Nijmegen.
Background and objective
In hemodialysis patients, deficiency of water-soluble vitamins can occur due to dietary restrictions and dialysis clearance. To prevent deficiency, vitamin supplementation is recommended by international guidelines. However, little is known about the optimal dosage of vitamin supplementation. Too much supplementation can result in toxic levels, which can result in peripheral neuropathy for vitamin B6, characterized by numbness, and tingling and burning sensations. Recently, we encountered peripheral neuropathy with toxic vitamin B1 and B6 levels in a series of hemodialysis patients. We designed this study to investigate the prevalence of high vitamin B1 and B6 levels in hemodialysis patients and to study the effect of dose reduction of supplementation on B1 and B6 levels.
Methods
This study was conducted in 27 patients from the Franciscus Gasthuis and Vlietland hospital, Rotterdam and the Catharina Hospital, Eindhoven. Vitamin B1 and B6 levels were assessed before and after dose reduction of supplementation (from once daily to twice or thrice weekly, after hemodialysis sessions). The primary outcome of the study was the percentage of patients with B6 levels outside target range (35-110 nmol/L) before and after dose reduction. Secondary outcomes were the median B1 and B6 levels before and after dose reduction, the percentage of patients with B1 levels outside target range (70-185 nmol/L) before and after dose reduction, plus the percentage of patients with B6 in the possible toxic range (> 500 nmol/L) before and after dose reduction. Statistical analysis was performed with SPSS version 28.0; B1 and B6 levels before and after dose reduction were compared with Wilcoxon signed rank test, differences in percentage within target range were assessed with the McNemar test.
Results
Before dose reduction, 85% of the patients had B6 levels outside the target range, versus 70% afterwards (P = 0.219). Median B6 levels decreased from 270 to 160 nmol/L (P < 0.001), B1 levels decreased from 231 to 192 nmol/L (P = 0.019) The percentage of patients within target range for B1 before and after dose reduction did not differ (33% and 30%, P = 1.00). Four (15%) and three (11%) patients had B6 levels > 500 nmol/L before and after dose reduction, respectively.
Conclusion
High vitamin B1 and B6 levels are extremely frequent in hemodialysis patients with daily vitamin supplementation. A dose reduction for vitamin supplementation in hemodialysis patients was effective in lowering vitamin B1 and B6 levels. We suggest to lower the dose frequency of supplementation to dosing only after hemodialysis sessions in clinical practice.
A complex, adherence-improving pharmacist intervention to reduce hyperphosphatemia in hemodialysis patients
F.J. van den Oever ab, E.C. Vasbinder a, T. van Gelder b, Y.C. Schrama c and P.M.L.A. van den Bemt d
a Department of Clinical Pharmacy, Franciscus Gasthuis and Vlietland, Rotterdam.
b Department of Clinical Pharmacology and Toxicology, Leiden University Medical Center.
c Department of Nephrology, Franciscus Gasthuis and Vlietland, Rotterdam.
d Department of Clinical Pharmacology and Pharmacology, University Medical Center Groningen.
Background and objective
Phosphate-binding drugs are used to treat hyperphosphatemia in hemodialysis patients, with unsatisfactory results in approximately half of the patients. A major problem is suboptimal treatment adherence, mainly due to forgetfulness, a high pill burden, a complex treatment scheme and demanding user instructions. Several pharmacist interventions have been developed to improve medication adherence to phosphate-binding drugs. Most of these were single, simple interventions with minor effects. Therefore, we designed a complex, adherence-improving intervention consisting of pharmacist consultations in which barriers to adherence were investigated and discussed, combined with a significant dose reduction of phosphate-binding drugs. We hypothesized that the complex intervention would lead to a reduction in mean phosphate concentrations of at least 0.25 mmol/L.
Methods
This was a prospective, intervention study in 69 hemodialysis patients with hyperphosphatemia (> 1.50 mmol/L) and a high pill burden of phosphate-binding drugs (≥ 6 tablets a day) in the Franciscus Gasthuis and Vlietland hospital in Rotterdam. The complex, adherence-improving intervention consisted of three pharmacist consultations with the patient at baseline, at 1-2 weeks, and at 3 months. At baseline the Quick Barrier Scan (QBS), to investigate barriers to adherence, and MARS-5 (patient-reported adherence) were administered. At 1-2 weeks, relevant subjects from the QBS were discussed; patient recommendations and a dose reduction for the phosphate-binding drugs were provided. After three months, a last consultation was performed to discuss patient experiences, and MARS-5 was repeated. The primary outcome parameter was the mean phosphate concentration in the three months after start of the intervention versus the three months before. Secondary outcome parameters were the pill burden for phosphate-binding drugs and patient-reported adherence (MARS-5). Data were analysed with SPSS version 28.0, a paired t-test was used to compare the mean phosphate concentrations and pill burden, the Wilcoxon signed rank test was used to compare MARS-5.
Results
The mean (± SD) phosphate concentration was 1.99 ± 0.38 mmol/L before versus 2.04 ± 0.35 mmol/L after the intervention (P = 0.193). Mean daily phosphate-binder pill burden decreased from 8.6 ± 3.1 units before to 5.7 ± 2.7 after the intervention (P < 0.001). Patient-reported adherence increased, although the median adherence did not change (median 24 IQR 22-25 before and median 24 IQR 23.25-25 afterwards, P = 0.008).
Conclusion
Although the intervention did not reduce phosphate concentrations, a major reduction in phosphate-binder pill burden was achieved. This may be due to improved adherence. This complex, adherence-improving intervention seems promising in decreasing pill burden and improving adherence, but our results need to be confirmed in larger, controlled studies.
The pharmacokinetic profile of olanzapine in anorexia nervosa patient: a case report
Kajie Liang a, Lisanne Krens a, Joris van der Vlugt b and Tessa Bosch ac
a Department of Hospital Pharmacy, Maasstad Hospital, Rotterdam.
b Antes - Parnassia group, psychiatric hospital.
c Department of Clinical Pharmacology & Toxicology MaasstadLab, Maasstad Hospital, Rotterdam.
Background and objective
Several studies have observed the efficacy of olanzapine in treating anorexia nervosa (AN) [1,2] , a condition characterized by severe underweight, affecting various pharmacokinetic processes. The absence of olanzapine pharmacokinetic studies in AN patients hinders treatment optimization. Clinicians often employ a conservative dosing approach, particularly in patients with the lowest BMIs, potentially leading to suboptimal treatment outcomes and a poorer prognosis. Our main objective is to investigate olanzapine's pharmacokinetics in AN patients to enhance precision dosing.
Methods
We present a case of a severely malnourished 38-year woman, diagnosed with anorexia nervosa per DSM-IV criteria. She required enteral feeding due to her restrictive eating disorder-induced malnutrition and deficiencies. To address anxiety, agitation, and anorectic obsession as the disease progressed, olanzapine was introduced and titrated from 2.5 mg to 10 mg once daily. We measured the olanzapine serum levels using a limited sampling method to determine the total drug exposure (AUC0-24h) and pharmacokinetic parameters at steady states.
Results
The BMI of the patient was restored from 12 (anorexic) at admission to 18.9 (normal) after six months of intensive psychotherapy, dietary interventions, and concurrent olanzapine pharmacotherapy. EDE-Q and Y-BOCS assessments showed clinical improvements of 87 and 19 scoring points, respectively, over this six-month period. In terms of olanzapine pharmacokinetics, the measured trough concentrations at steady state were 6, 4, and 16 μg/L for daily doses of 2.5 mg, 5 mg, and 10 mg, respectively. Drug clearance values were 7.5, 10.2, and 11.6 L/hr, the volume of distribution was 11.3, 12.8, and 16.5 L, and the calculated total exposures (AUC0-24h) were 200.5, 147.3, and 517.5 μg/L*h, with tmax values of 4, 6, and 6 hours, and half-lives of 22.2, 15.1 and 20.1 h were for the respective doses.
Conclusion
Since there is no established therapeutic range for anorexia nervosa, we can only observe that the trough concentrations are relatively lower compared to the therapeutic levels (20-80 μg/L) seen in schizophrenia treatment. The patient tolerated the 10 mg olanzapine dosage well, resulting in a trough level of 16 μg/L. Further research is necessary to better understand the pharmacokinetics in this population and establish a therapeutic range for anorexia nervosa, thus enabling optimized precision dosing in this vulnerable patient group.
Alterations of appetite-regulating hormones in risperidone treated children and adolescent - A posthoc analysis of the SPACe study
J. Liang a, B.C.M. de Winter ac, R.A. Hermans b, S.M. Kloosterboer b, I. Bayraktar a, M.H.J. Hillegers b, S.A.A. van den Berg e, B.C.P. Koch c and B. Dierckx b
a Department of Hospital Pharmacy, Erasmus University Medical Center, Rotterdam.
b Department of Child and Adolescent Psychiatry/Psychology, Erasmus University Medical Center, Rotterdam.
c Rotterdam Clinical Pharmacometrics Group, Erasmus University Medical Center, Rotterdam.
d Department of Pharmacy, Faculty of Pharmacy, Hacettepe University, Ankara, Turkey.
e Department of clinical chemistry, Erasmus MC, University Medical Center Rotterdam.
Background and objective
Risperidone effectively reduces irritability and hyperactivity over the short and long term. However, its benefits are hindered by metabolic consequences like excessive weight gain and cardiometabolic issues, posing higher risks in children than adults. Metabolic syndrome during childhood is a strong indicator of future risks like type 2 diabetes and cardiovascular disease in adulthood. Previous studies have shown a dose-dependent effect of risperidone on weight gain and metabolic changes, establishing a link between risperidone exposure and weight gain. This study explores the impact of risperidone on appetite-regulating hormones (e.g., insulin, leptin, bioleptin) and their relation to change in body weight over time, aiming to better understand and prevent long-term side effects of antipsychotic-induced weight gain and metabolic issues.
Methods
We analysed data from ten patients participating in the SPACe study, a multicentre cohort in the Netherlands. These patients, aged 6-18 years, starting risperidone treatment for autism spectrum disorder, were monitored for risperidone and hormone levels over a 6-month period, as detailed by Kloosterboer et al. [1]. We aim to investigate the impact of risperidone treatment on these hormones (e.g., insulin, leptin, bioleptin) by assessing the changes over time. We use scholar-paired t-test to evaluate changes in HOMA-IR index between baseline and 24 weeks, and the Wilcoxon two-tailed signed rank test was used to assess the alteration in leptin and bioleptin. With linear logistic regression we attempt to predict the correlation of the appetite-regulating hormones and risperidone exposure.
Results
Ten patients (80% male, median age 9.7 year, and median body weight 32.8 kg) were included. Statistical comparison of baseline, and 6-month measurements, showed a significant increase in bioleptin (P < 0.05) along with BMI z-score. Significant correlations between the appetite-regulating hormones and BMI z-score were observed at baseline but disappeared following risperidone treatment, suggesting an altered association. Statistically significant correlations with the risperidone exposure (sum trough concentration) appeared for leptin and bioleptin, and insulin (P < 0.05) shows a declining trend (P < 0.1).
Conclusion
In this study, we have noted increasing trends in serum leptin, bioleptin, insulin levels, and HOMA-IR index, which correspond to BMI-z at baseline but to a lesser extend following risperidone treatment. Moreover, significant inverse correlations between leptin, bioleptin, and risperidone exposure were observed. These findings warrant further investigation. In the upcoming SPACe2: STAR follow-up study, we will explore these relationships in a larger cohort.
Drug waste of ready-to-administer syringes in the Intensive Care Unit: aseptically prepared syringes versus prefilled sterilized syringes
Thomas G. van Gelder ab, Arief Lalmohamed ac, Kim D. Dorst-Mooiman b, Jan C. Dekker b, Marcel J. Schinkel a, Maaike A. Sikma d, Esther V. Uijtendaal a and Toine C.G. Egberts ac
a Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University.
b Department of Clinical Pharmacy, Drug Production & Compounding Unit, University Medical Center Utrecht, Utrecht University.
c Division of Pharmacoepidemiology & Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
d Intensive Care and Dutch Poisons Information Center, University Medical Center Utrecht, Utrecht University.
Background
The availability of ready-to-administer (RTA) syringes for intravenous (IV) drugs facilitates rapid and safe administration in emergency and intensive care situations. Hospital pharmacies can prepare RTA syringes through aseptic batchwise filling. Due to excess production of these RTA syringes for sufficient availability for patient care and their limited (microbiological) shelf life, waste is unavoidable, which contributes to environmental pollution. RTA prefilled sterilized syringes (PFSSs) have much longer shelf lives than aseptically prepared RTA syringes and might contribute to reducing drug waste.
Methods
We measured drug waste of RTA syringes over an 8-year time period from August 2015 to May 2023 in the 32-bed ICU of the University Medical Center Utrecht. We distinguished between RTA syringes prepared through aseptic batchwise filling by our hospital pharmacy (“RTA aseptic syringes”, shelf life of 31 days) and RTA PFSSs (shelf life of 18 months). An intervention group of three drug products that were replaced by PFSSs was compared to a control group of five drug products that were not replaced by PFSSs during the study period. We then defined four different periods within the total study period, based on quarantine time of the RTA aseptic syringes and time of PFSS introduction: (1) no quarantine, (2) 3-day quarantine, (3) 7-day quarantine and (4) PFSS introduction. Our primary endpoint was the number of RTA syringes that was wasted, expressed as the percentage of the total number of syringes dispensed to the ICU in each of these four periods. We used a Kruskall-Wallis test to test if waste percentages differed between time periods in the control and intervention groups, with a post-hoc Dunn’s test for pairwise comparisons. Furthermore, we applied two interrupted time series (ITS) analyses to visualize and test the effect of introducing different quarantine times and the PFSSs on waste percentage.
Results
Introduction of PFSSs significantly decreased drug waste of RTA syringes irrespective of drug type in the intervention group, from 31% during the 7-day quarantine period to 5% after introduction of the PFSS (P < 0.001). The control group showed no significant decrease in drug waste over the same time periods (from 20% to 16%; P = 0.726). We observed a significant difference in the total drug waste of RTA aseptic syringes between time periods, which may be attributed to the implementation of different quality control quarantine procedures.
Conclusion
Drug waste of RTA syringes for the ICU can be significantly decreased by introducing PFSSs, supporting hospitals to enhance environmental sustainability. Furthermore, the waste percentage of RTA syringes prepared through aseptic batchwise filling is significantly impacted by duration of quarantine time.
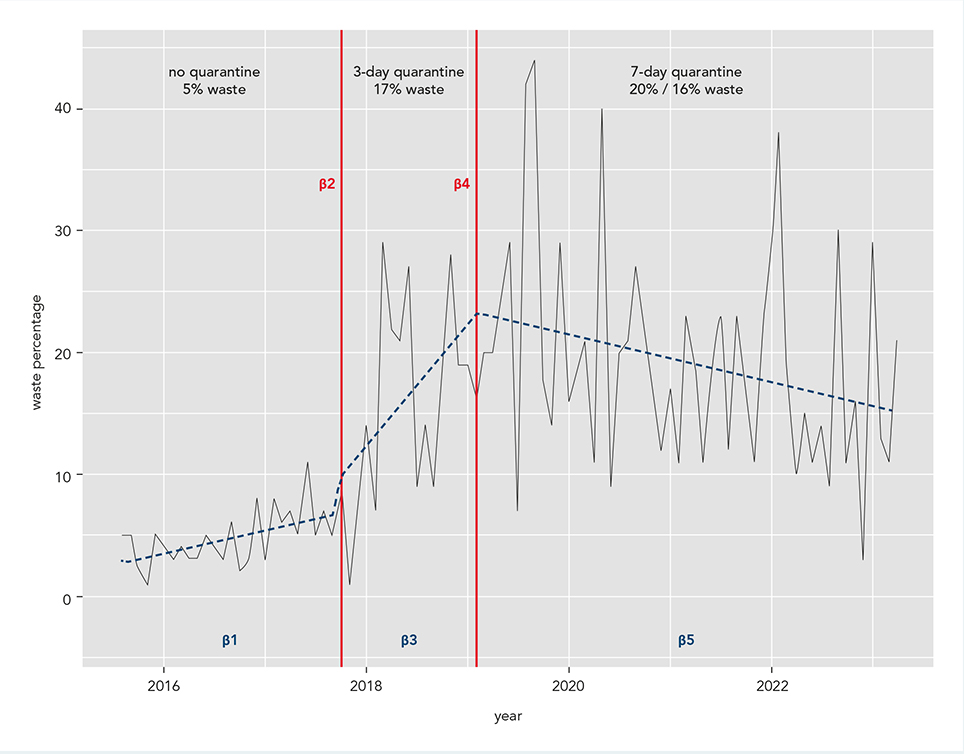
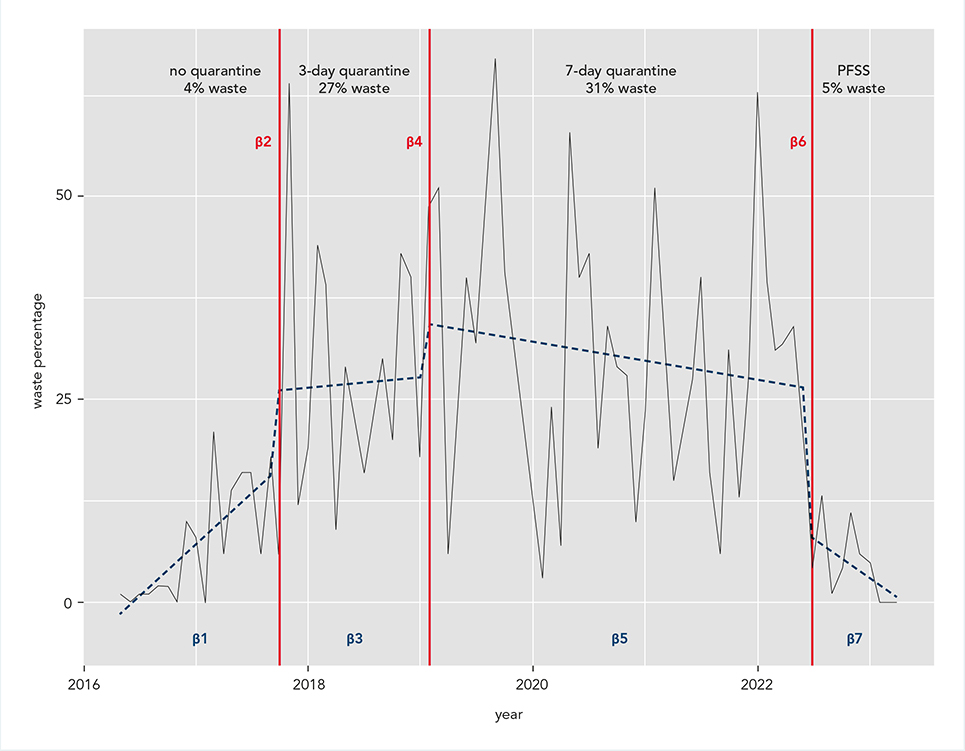
Redispensing of expensive oral anticancer medicines: A practical application
Kübra Akgöl a, Lisanne N van Merendonk a, Hannerieke J Barkman a, Dorieke EM van Balen a, Hester L van den Hoek a, Marjolein G Klous a, Jeroen JMA Hendrikx ab, Alwin DR Huitema acd, Jos H Beijnen a and Bastiaan Nuijen a
a Department of Pharmacy and Pharmacology, The Netherlands Cancer Institute-Antoni van Leeuwenhoek, Amsterdam,
b Department of Nuclear Medicine, The Netherlands Cancer Institute-Antoni van Leeuwenhoek, Amsterdam.
c Department of Pharmacology, Princess Máxima Center for Pediatric Oncology, Utrecht.
d Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University.
Background
Increasing use of expensive oral anticancer medicines comes with the downside of a financial and environmental burden, partially caused by unused medication. Returned oral anticancer medicine to the pharmacy could be considered for redispensing providing guaranteed quality.
Objective
This study aimed to identify and implement quality aspects and criteria for redispensing oral anticancer medicine in daily pharmacy practice.
Methods
A systematic analysis was conducted to determine the eligibility of oral anticancer medicine for redispensing. Over a one-year period, the number of returned oral anticancer medicine accepted for redispensing was quantified, and the reduction in financial waste and environmental burden calculated based on this assessment.
Results
Four categories of quality aspects were identified for determining the eligibility of oral anticancer medicine for redispensing (figure 1): product presentation suitability (stability characteristics, storage requirements), physical condition (unopened or opened secondary or primary packaging, visual appearance), authentication (Falsified Medicines Directive, confirmation of initial dispense, recall), and additional aspects (remaining shelf life, period of storage in uncontrolled conditions). A standardized procedure for redispensing was implemented in daily pharmacy practice. During the study period, 10,415 oral anticancer medicine dose units out of 13,210 returns (79%) were accepted for redispensing. The total value of oral anticancer medicine accepted for redispensing was € 483,301, accounting for 0.9% of the total value dispensed during this period. Furthermore, the potential reduction in environmental burden was estimated at 1132.1 g of potent active pharmaceutical ingredient.
Conclusion
By implementing strict procedures considering all relevant quality aspects, redispensing of oral anticancer medicine can be successfully implemented into daily pharmacy practice, resulting in a significant reduction in financial waste and environmental burden.
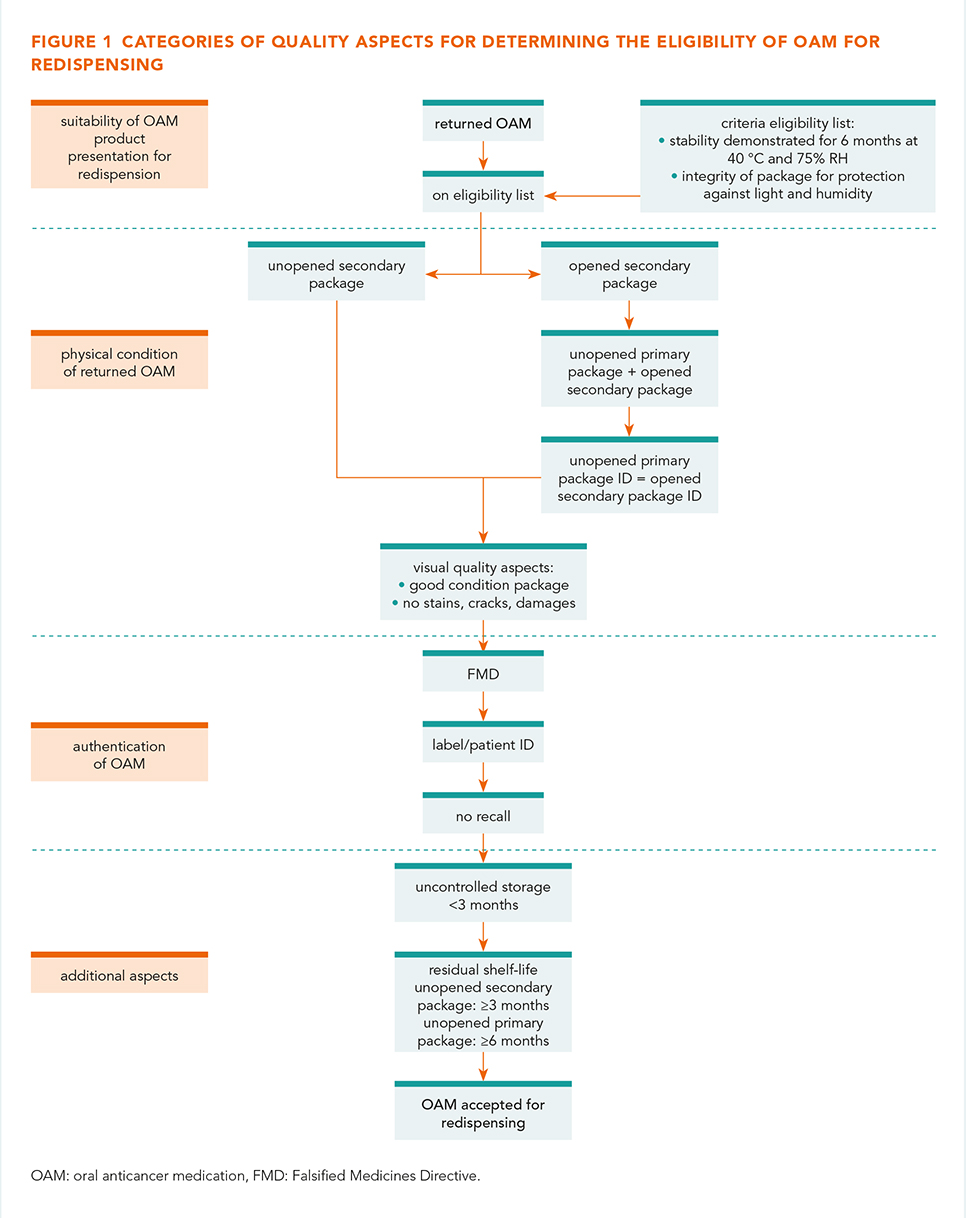
Stability of piperacillin/tazobactam for continuous infusion improved by addition of sodium citrate
K. Stender a, E.M. Kemper a, A.L. Lindemans b and C. Oussoren a
a Apotheek A15, Gorinchem.
b Erasmus University Medical Center, Rotterdam.
Background
Piperacillin instability is the limiting factor for the antimicrobial treatment of patients at home with intravenous piperacillin/tazobactam. In the past, outpatient treatment was possible by using the licenced medicine Tazocin. This medicinal product contained citric acid and disodium edetate for sufficient stability after reconstitution. However, Tazocin is no longer available in the Netherlands. The available generic products do not contain any excipients, which means that the shelf life after reconstitution is limited to 2 days refrigerated and 8 hours room temperature.
Objective
The goal of our study was to investigate whether addition of a sodium citrate buffer to the infusion solution could improve the stability over a prolonged period of time.
Methods
The stability of piperacillin and tazobactam with sodium citrate has been studied at two concentrations. The infusion solutions were prepared by the hospital pharmacy of Erasmus MC, containing piperacillin/tazobactam 10/1.25 g and 45/5.63 g in 500 mL saline 0.9% (both concentrations n = 2). The content of each vial of piperacillin/tazobactam 4/0.5 g was dissolved with 17.3 mL of sodium citrate solution 40 mg/mL, which was developed by Apotheek A15. To mimic the home infusion situation, the infusion bags were kept for 15 days of which 13 days refrigerated (2-8 °C) followed by two days at room temperature (15-25 °C).
Analysis was performed by the Laboratorium der Nederlandse Apothekers (LNA). High-performance liquid chromatography was used for assessing the concentration of piperacillin and tazobactam. Also the pH, colour and clarity of the solutions were measured. Stability has been studied at day 0, 3, 7, 13, 14 and 15.
Results
Concentrations of piperacillin and tazobactam, for both the low and high concentrations, hardly change during the period of study (see table 1 and figure 1). A slight decrease of 6.8 to 6.4 in the pH was observed in all solutions tested, however, the pH remained within the stability optimum of piperacillin (5.5-7.0). No crystallization or visible changes in colour and clarity of the solutions were observed.
Conclusion
Our study showed that addition of sodium citrate to piperacillin-tazobactam solution leads to an improved stability of piperacillin. Reconstitution of piperacillin/tazobactam 4/0.5 g vials with 17.3 mL sodium citrate solution 40 mg/mL results in a relatively stable solution. Stability has been demonstrated for 15 days, consisting of 13 days in the refrigerator, followed by 48 hours at room temperature. The extended shelf life simplifies the logistics surrounding home treatment of piperacillin/tazobactam-sensitive microorganisms in the Netherlands.
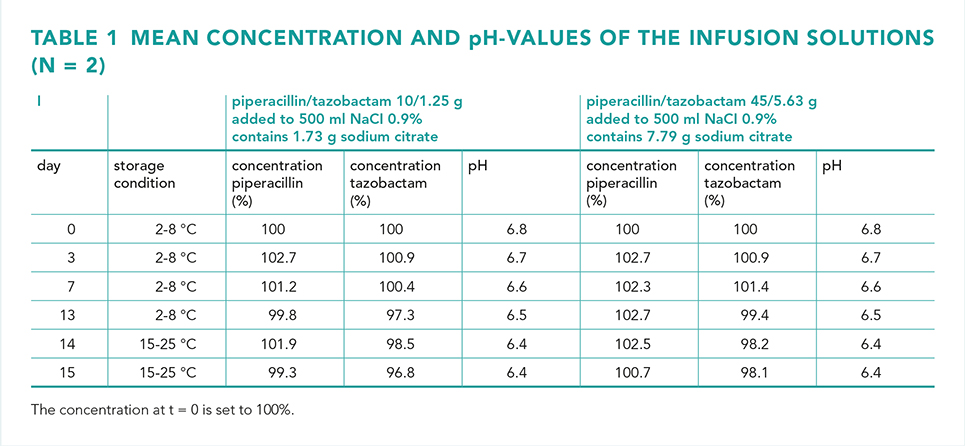
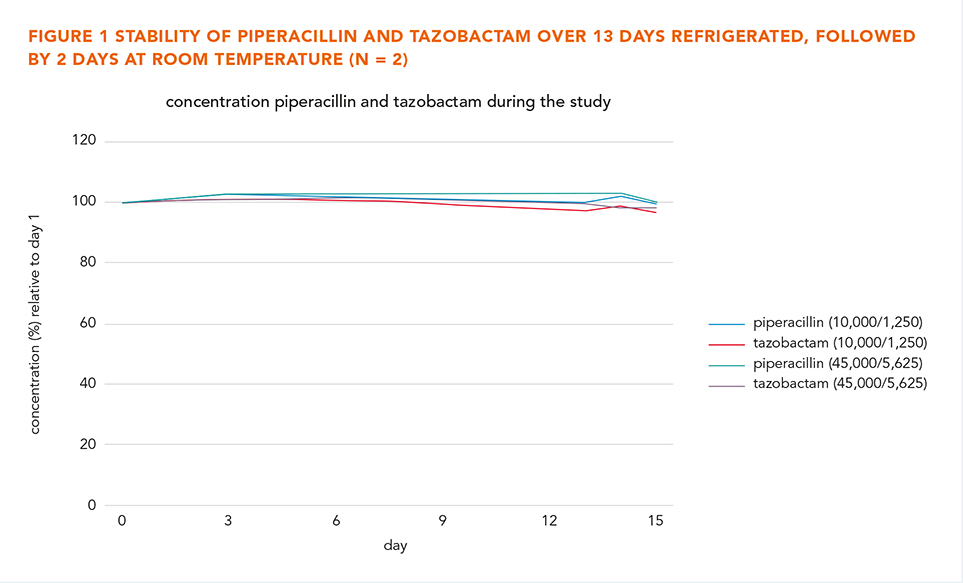
Relative bioavailability of dolutegravir (DTG) and emtricitabine/tenofovir alafenamide fumarate (FTC/TAF) administered as paediatric tablet formulations in healthy volunteers
Lisanne Bevers a, Anne Kamphuis a, Wendy van der Wekken-Pas a, Rory Leisegang b, Linda Lewis c, David Burger a and Angela Colbers a
a Department of Pharmacy, Radboud Institute for Medical Innovations (RIMI), Radboud University Medical Center, Nijmegen.
b Clinical Pharmacology, Paediatric Centre of Excellence, Gilead Sciences, Dublin, Ireland.
c Clinical and Regulatory Affairs, Product Development and Regulatory Affairs team, Clinton Health Access Initiative (CHAI).
Background and objective
In the EDCTP2-funded UNIVERSAL project (RIA2019PD-2882) a paediatric fixed dose combination product containing dolutegravir/emtricitabine/tenofovir alafenamide (DTG/FTC/TAF) will be developed. However, data on co-administration of paediatric tablets for oral suspension (TOS) are currently lacking. We undertook a relative bioavailability (RBA) study in healthy volunteers to investigate a potential pharmacokinetic effect when paediatric DTG and FTC/TAF as TOS are taken together.
Methods
A single dose, single-centre, open-label, 3-period, randomised, cross-over RBA study in healthy adult volunteers was conducted at the Radboud university medical center, Nijmegen, the Netherlands. Participants received the following treatments in randomized order: single dose of 3x60/7.5 mg (180/22.5 mg) FTC/TAF TOS (treatment A); single dose of 6x5 mg (30 mg) DTG TOS (treatment B); single dose of 180/22.5 mg FTC/TAF plus 30 mg DTG TOS (treatment C). Blood samples were collected at time = 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1.0, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24 and 48 hours post-dose. Plasma concentrations of all compounds, including the pharmacologically active tenofovir (TFV), were measured using a validated LC-MS/MS method. We applied the statistical method used in bioequivalence studies with the following pre-defined criteria: if the 90% CIs of the Geometric Least Squares Mean (GLSM) ratios of AUC and Cmax of each compound in treatment C relative to treatment A or B is within 0.70-1.43, a potential clinically relevant pharmacokinetic interaction in treatment C was ruled out.
Results
15 healthy volunteers were included. GLSM ratios (90% CI) were 1.02 (0.94-1.10) for DTG AUC0-∞ and 1.16 (1.10-1.23) for DTG Cmax; 0.84 (0.70-1.01) for TFV AUC0-last and 1.06 (0.92-1.23) for TFV Cmax; 0.97 (0.92-1.02) for FTC AUC0-∞ and 1.05 (0.94-1.17) for FTC Cmax. For TAF the lower boundary of the 90% CI of the GLSM ratio was below 0.70 for AUC0-∞ (0.62) and Cmax (0.65). However, only half of the participants had an individual ratio below 1 and half above 1, likely due to high inter-subject variability. Moreover, TAF is a prodrug of TFV, and no relevant interaction was found for the latter.
Conclusion
We found no clinically relevant pharmacokinetic interaction between DTG TOS and F/TAF TOS for the compounds DTG, TFV, and TFV. For TAF the AUC0-∞ and Cmax 90% CIs of the GLSM ratios fell outside our pre-defined criteria. However, no consistent effect was found and TFV exposure is considered clinically more important than TAF exposure.
Is newer always better? A quality and cost comparison of compounded capsules versus 3D printed tablets
S. Ayyoubi, R.A.Ramkisoen, Z. Kafa Z and E.J. Ruijgrok
Erasmus MC, Erasmus University Medical Center Rotterdam, Department of Clinical Pharmacy.
Background
Compounding of medication is of crucial importance for providing essential individual care. However, it can come with quality risks because of small-scale production processes and less stringent regulations compared to registered medication. Several studies have demonstrated quality defects of compounded capsules. One study analysed compounded hydrocortisone capsules and found that 21% of the batches did not demonstrate content uniformity (CU) and 4% did not even contain hydrocortisone. 3D printing (3DP) is an automated technology which allows pharmacists to produce medication tailored to the needs of the individual patient. With 3DP, it is possible to produce high-quality medicines where shape, dose, release-profile and taste can be personalized.
Objective
To support decision-making, and integration of 3DP in pharmaceutical care, we aim to assess the quality and costs of 3D printed hydrocortisone tablets, versus compounded hydrocortisone capsules.
Methods
Three batches of 10 mg hydrocortisone tablets were printed using semi-solid extrusion 3DP (SSE). Manually filled 10 mg hydrocortisone capsules were purchased from three compounding pharmacies (CP) and one batch was filled by a pharmacy student (PS). Hydrocortisone content (n = 10) and release profiles (n = 6) were determined for each formulation.
Results
SSE batches are closest to target, demonstrated by CU results (figure 1), also confirmed by acceptance values (AV) (table 1), where AV < 15 is a requirement.
Dissolution study results show that all formulations comply to compendial standards of 80% release < 1 hour, although 2 SSE tablets were closest to the 10 mg target again (figure 2).
Conclusion
Preliminary data show that SSE is a feasible method to produce direct release hydrocortisone tablets with a higher quality compared to compounded capsules. SSE tablets and capsules were fabricated by a PS, demonstrating that 3D printing is an easier method for unexperienced personnel to produce high quality products compared to capsule filling. Pharmacists should embrace 3DP to make their added value visible by utilizing it to produce personalized medication, a skill that distinguishes pharmacists from other professions. Data from other CP capsules are yet to be gathered and a cost-comparison analysis is on the way.
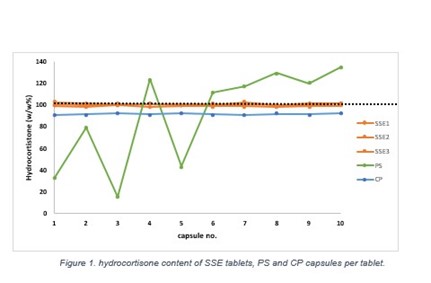
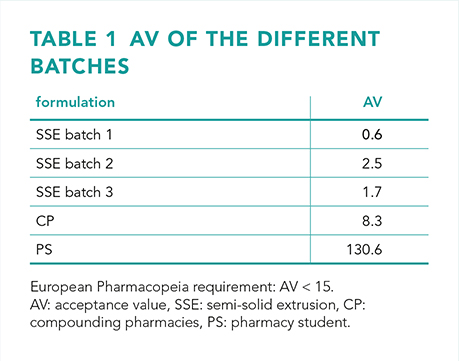
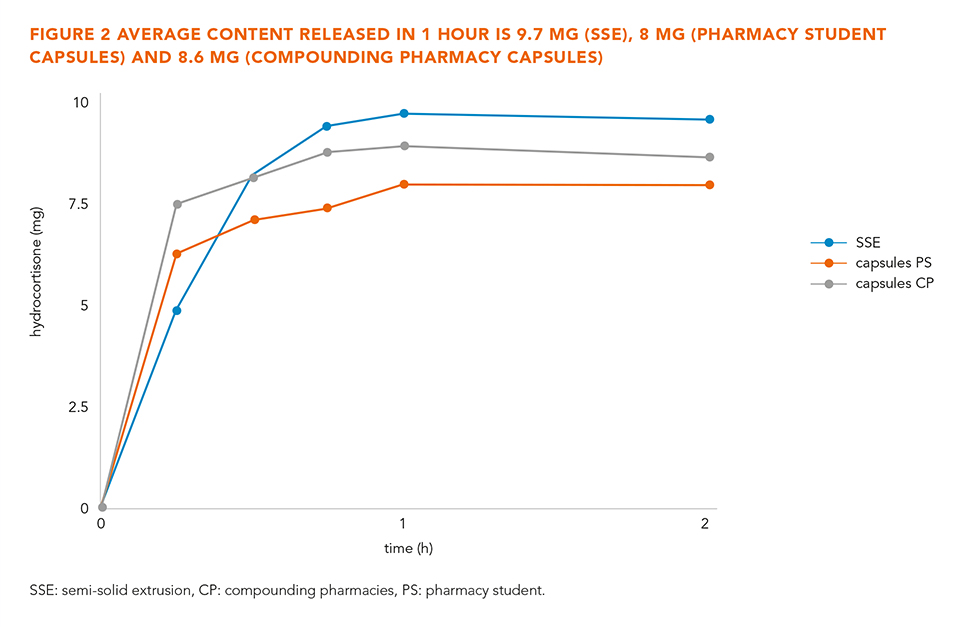
Compounding with glass ampoules in Dutch hospitals: survey results
H. Şahin ab†, A. Singh bc†, H. Abdullah-Koolmees bd and F. Karapinar-Çarkit abc
a Department of Clinical Pharmacy and Toxicology, MUMC+ Hospital, Maastricht.
b Utrecht Pharmacy Practice network for Education and Research, Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
c Department of Clinical Pharmacy, OLVG Hospital, Amsterdam.
d Department of Pharmacy & Clinical Pharmacology, Amsterdam University Medical Center, Vrije Universiteit Amsterdam.
†These authors contributed equally to this work and share the first authorship.
Background and objective
Glass ampoules find extensive application in healthcare for intravenous administration, pulmonary nebulization, and oral preparations (e.g., caffeine). Dutch compounding guidelines recommend the use of filter needles or straws during the handling of glass ampoules [1]. Compliance in pharmacy departments and on wards remains uncertain. Hence, the primary objective of this study was to evaluate filter needle/straw use, glass particle observation, and ampoule disposal due to particle contamination among pharmacy technicians and nurses. In addition, we assessed the handling of glass ampoules during medication procurement in hospital pharmacies.
Method
The study employed an observational approach, utilizing a questionnaire developed by the hospital pharmacy section of UPPER (pharmacy practice research of Utrecht University). The questionnaire focused among others on the management of glass particles and policies in the procurement of glass ampoules. Data collection occurred during master pharmacy students' hospital pharmacy internships between September and November 2022. These students conducted interviews with pharmacists, pharmacy technicians (both within the pharmacy and on hospital wards), and nurses. Descriptive data analysis was used.
Results
Students obtained data from 31 Dutch hospitals (6 academic, 15 top clinical and 10 peripheral). In the context of hospital pharmacy compounding, 14% of hospitals did not employ a filtering technique (except for intrathecal preparations). For pharmacy technicians working on hospital wards, 23% of hospitals did not use filtering techniques, and this figure increased to 50% for nurses (unregularly implemented). Students interviewed 50 pharmacy technicians in the hospital pharmacy, 51 technicians on wards, and 50 nurses. Results showed that 82% of pharmacy technicians in the hospital pharmacy reported encountering glass particles during compounding, which increased to 92% on wards. Additionally, 45% of nurses observed glass particles. In the hospital pharmacy, 16% of pharmacy technicians reported discarding ampoules due to glass particles, compared to 19% on wards and 20% among nurses. Only nine hospital pharmacies had established policies to mitigate the use of glass ampoules during medication procurement.
Conclusion
The research revealed variations in the adoption of filtering techniques for glass ampoules across different hospitals. Hospital pharmacies predominantly implemented these techniques. Both pharmacy technicians and nurses reported instances of observing glass particles in the ampoules, leading to the disposal of such ampoules. Future studies should investigate the underlying causes of disparities between pharmacy departments and hospital wards. Moreover, further studies are warranted to evaluate the potential health consequences of glass particle exposure.
Reference
1. KNMP. LNA-procedures Productzorg - Aseptische handelingen. [Internet]. Available from: https://kennisbank.knmp.local/article/LNA-procedures_productzorg/asep/F114.html. [Accessed 31st August 2023].
Impact of longitudinal interprofessional education on pharmacy and medical students’ competence development
J.F. Mertens a, M.H.M. Hessel a, K. Kroeze a, C.W. Walinga a, A.F. Norbart a, V.H.M. Deneer b, T.G.H. Kempen b, E.S. Koster b, M.L. Bouvy b and T. van Gelder a
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
b Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences (UIPS), Department of Pharmaceutical Sciences, Utrecht University.
Background
Collaborative relationships between pharmacists and physicians are essential for optimizing pharmaceutical patient care. To ensure that medical and pharmacy graduates are competent to collaborate, interprofessional education (IPE) is stimulated. However, evidence is lacking on the superiority of IPE to uniprofessional education (UPE) and IPE is often limited to a single educational activity. Three pharmacotherapy-related mandatory activities are designed as part of a longitudinal program throughout the master curricula of pharmacy and medicine at Leiden University Medical Center. Activities may be interprofessional or uniprofessional, since medical students outnumber pharmacy students. This study aims to determine the impact of this longitudinal IPE pharmacotherapy program on pharmacy and medical students’ perceived competence development.
Methods
The Interprofessional Collaborative Competency Attainment Scale (ICCAS), a validated 20-item self-report instrument to score perceived development of interprofessional core competencies, was given to 461 students of 17 educational activities within the program between September 2022 and June 2023. Study participants rated how competent they felt before and after the learning activity for each competency, anonymously. Paired student’s t-tests and effect sizes using Cohen’s d were calculated. Subgroup analysis evaluated the impact of multiple activities completed in the program.
Results
In total, 294 surveys were completed (response rate 64%). All 20 ICCAS items improved significantly with UPE and IPE. Significantly larger effect sizes were found with IPE especially in the categories of collaboration, roles and responsibilities, and team functioning. These effects increased significantly with the number of IPE activities. The categories with the least robust effect sizes were communication and conflict management.
Conclusion
Participation in multiple IPE activities increases interprofessional competence development more than UPE and a single IPE activity. These findings stimulate the implementation of a longitudinal IPE program into medical and pharmacy curricula and can be used to improve existing educational activities and expand to new activities to address other interprofessional competencies. In our follow-up study, we focus on gaining a deeper understanding of how multiple IPE activities influence students’ perceptions of collaboration in clinical practice.
Assessing the impact of drug recalls on patients in the Netherlands: a 5-year retrospective data analysis
P.A. Annema ab, H.J. Derijks a, M.L. Bouvy c and R.J. van Marum ab
a Department of Pharmacy and Clinical Pharmacology, Jeroen Bosch Hospital, ‘s-Hertogenbosch.
b Department of Elderly Care Medicine, Amsterdam Public Health Research Institute, Amsterdam UMC, Location VUmc.
c Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences (UIPS), Faculty of Science, Utrecht University.
Background and objective
Drug recalls occur frequently and have the potential to impact considerable numbers of patients and healthcare providers. However, none of the drug regulatory agencies could provide a readily accessible overview of drug recalls. Therefore, the extent of conducted recalls and their impact on patients remains unknown. To address this, we developed a comprehensive overview of drug recalls affecting patients and estimated how many patients in the Netherlands had to switch their medications as a result.
Methods
We compiled this overview based on the drug recall registrations from the Jeroen Bosch Hospital (JBZ), the University Medical Center Utrecht (UMCU), and the Royal Dutch Pharmacists Association (KNMP). A retrospective data analysis was conducted to identify drug recalls that affected patients. Specifically, we defined these as drug recalls that required patients to actively switch their drug to a different batch or brand of the same drug or to switch to a drug within the same or a different class of drugs. To quantify the impact of these recalls we used drug dispensing data. Several drug recalls were carried out simultaneously, pertaining to the same drug type, with identical product defects, and affecting the same group of patients. In these cases, we combined drug dispensing data from these identical drug recalls and presented active drug users per patient population affected by (a series of) drug recalls.
Results
Between January 1, 2017, and December 31, 2021, we identified 48 drug recalls that impacted 35 patient populations and necessitated an estimated 855,000 patients to make active changes to their medications (figure 1). Among these recalls, those arising from concerns of product contamination and sterility issues had the most significant impact, affecting 630,000 patients. Most of the affected patients (292,000) were required to switch to a different brand of the same drug, whereas 95,000 patients had to switch to another drug class, as depicted in figure 2.
Conclusion
This overview of drug recalls affecting patients revealed that over a 5-year period, an estimated 855,000 patients had to switch their drugs. Future research should focus on elucidating the experiences of patients, but also physicians, pharmacists, and other stakeholders with drug recalls as well as ascertaining patient and healthcare provider preferences concerning drug recalls.
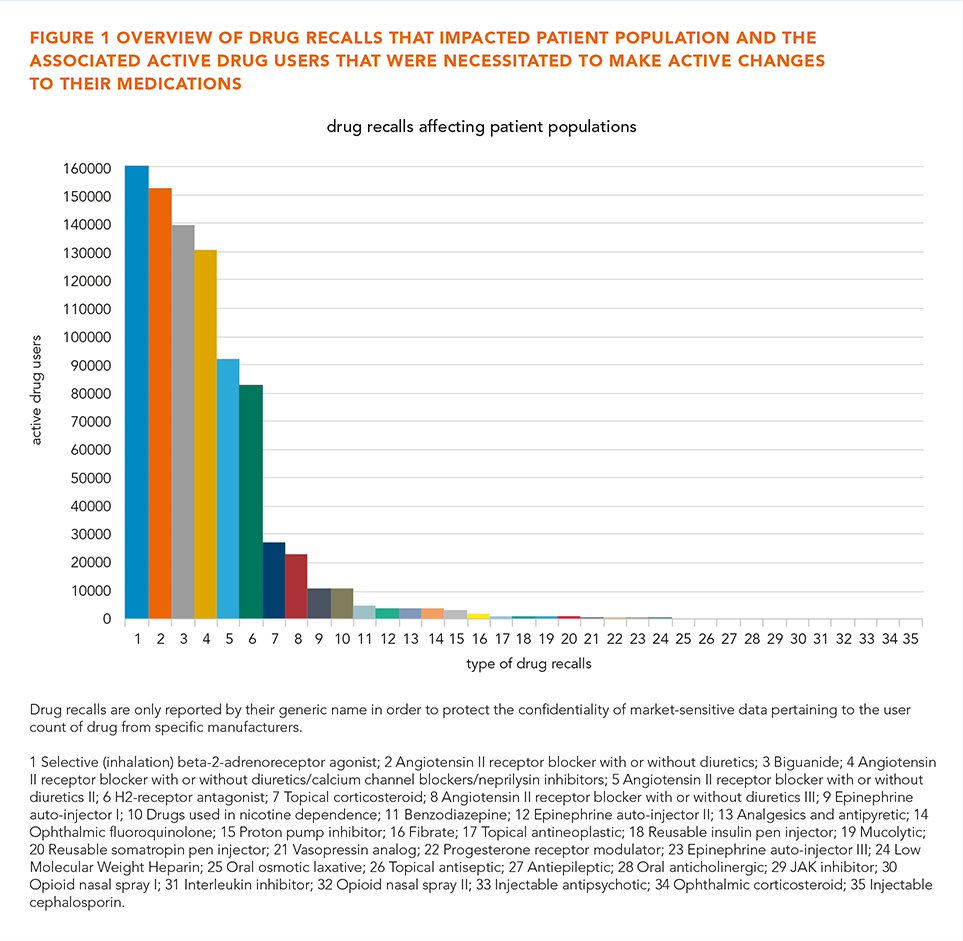
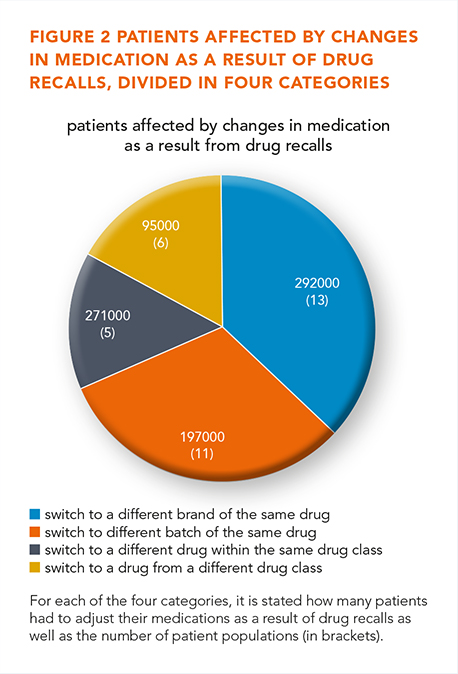
Performance of ChatGPT to questions regarding clinical pharmacy
Merel van Nuland a, Abdullah Erdogan a, Cenkay Aςar a, Ramon Contrucci b, Sven Hilbrants c, Lamyae Maanach d and Paul D. van der Linden a
a Department of Clinical Pharmacy, Tergooi Medical Center, Hilversum.
b Department of Clinical Pharmacy, Amphia Hospital, Breda.
c Department of Clinical Pharmacy, Leeuwarden Medical Center.
d Department of Clinical Pharmacy, Haga hospital, The Hague.
Background
ChatGPT is a language model that was trained on a large dataset encompassing a wide array of topics, including medical literature. Several studies have described the performance of ChatGPT on medical exams.
Objective
The main objective is to study its performance in answering exam questions regarding clinical pharmacy.
Methods
Questions were obtained from a Dutch application called ‘parate kennis’. This application features multiple choice questions to maintain a basic knowledge level in clinical pharmacy for hospital pharmacists and residents. Each question was presented to ChatGPT and its responses were evaluated for accuracy, concordance, quality of the substantiation and reproducibility. Quality of the substantiation was graded by two independent pharmacists using a 4-point scale, and a senior hospital pharmacist was consulted in cases of disagreement. Reproducibility was established by presenting questions multiple times and on various days. Accuracy results were compared to the overall score by pharmacists over 2022.
Results
A total of 264 clinical pharmacy-related questions were included in the analysis. ChatGPT yielded accurate responses for 79% (209/264) of the questions, surpassing pharmacists' accuracy of 66% (174/264). Notably, ChatGPT excelled in the fields of oncology, nephrology, and psychiatry, while its performance was less favourable in compounding, pharmaceutical analyses, neurology, and geriatrics. Concordance was 95% (252/264) and the quality of substantiation was deemed good or excellent for 72% (190/262) of the questions. Reproducibility was consistently high, both within-day and between-days (> 90%), as well as across different users (100%).
Conclusion
ChatGPT demonstrated a high accuracy and reproducibility to questions related to clinical pharmacy. Nonetheless, its performance falls short of meeting the requisite standards for patient care. Consequently, we posit that ChatGPT could serve as a valuable resource to pharmacists. We hope the technology will further improve, which may lead to enhanced future performance.
Bridging the gaps in hospital medication supply: implementation of an autonomous delivery robot - a user case and financial overview
F. van Tienen, C. Badia Lopez, M.J. Rutten-van Kranenburg and E.J. Ruijgrok
Erasmus MC, Erasmus University Medical Center Rotterdam, Department of Clinical Pharmacy.
Background and objective
A shortfall of thousands of healthcare professionals is expected in the next decade. Given the resulting increasing workload, digital health technology is being recognized as a means to enhance efficiency and safety in drug storage, dispensing and administration. Notable innovations in this area include the utilization of barcodes in medication administration, automated compounding, automated unit dose dispensing systems (ADDS) and autonomous delivery robots (ADR).
The Clinical Pharmacy at Erasmus MC has implemented an ADR as a next step device mounting on the tremendous gains of the ADDS and the pneumatic tubing system. In this pilot, the ADR is employed on two routes where previously logistic pharmacy personnel was doing the transportation rounds. This research provides a cost benefit analysis of the implementation. Additionally, it highlights both the (non)financial risks and opportunities of this innovation.
Methods
The implementation of the Relay Robotics ADR (Swisslog) was studied. In the 5-week pre-implementation phase, the average daily number and duration of deliveries to two oncology daycare wards was recorded. Initial investments needed for adequate functioning and communication of the ADR were assessed. Post-implementation, the same data were collected and compared to the pre-implementation phase, furthermore a qualitative assessment on the acceptance and experiences of people encountering the ADR was performed.
Results
Logistics personnel spent an average of 8.25 hours per week (approximately 0.25 FTE) making deliveries to oncology day treatment departments. Initial investments were very labour-intensive and took several months. The work consisted of installation of hardware in automatic doors and elevators, adjustments to networking infrastructure and providing the ADR with a new outer design to align with Erasmus MC house style. During the implementation phase, some technical problems were encountered, these were all resolved. After implementation, more than 95% of all deliveries were successfully taken over by the ADR, reducing the amount of hours spent by personnel to almost 0. Final cost calculations will be done in October 2023. The qualitative assessment yielded mostly positive reactions, however some negative emotions were expressed regarding the potential loss of jobs.
Conclusion
The implementation of an ADR at Erasmus MC was time and resource consuming but successful. The ADR takes over tasks from pharmacy staff so they can be deployed elsewhere in the pharmacy. In general, people’s reactions to the ADR were very positive. This research underlines the potential of automation for the relief of health care professionals in times of staff shortage.
Drug shortages in a Dutch hospital pharmacy: an overview
S. Taspinar and S.W. Zielhuis
Reinier de Graaf Gasthuis, Delft.
Background
The shortage of essential drugs is an increasing problem that poses a significant challenge for pharmacies and healthcare providers worldwide, as well as for the Netherlands. The Royal Dutch Pharmacists Association (KNMP) reported a total of 1514 drug supply disruptions for 2022, compared to 1007 in 2021. However, the impact of drug shortages in Dutch hospitals has not been properly investigated and described yet. Therefore, we analysed and categorized drug shortages in Reinier de Graaf Gasthuis hospital in Delft for a certain period.
Methods
A web-based system of forms was created to monitor all changes in the supply of the hospital pharmacy. Each change in a period of two years (01-01-2021 till 31-12-2022) was noted in either a temporary drug shortage, permanent drug shortage (due to discontinuation on the market), change due to an improved packaging option, cheaper alternative, a new contract with another supplier, a switch back to the original drug, or due to any other reason. After this period, data could be extracted.
Results
A total of 294 drug shortages were reported in 2021 and this amount increased to 371 in 2022. 114 shortages were permanent (from the total amount of 665 in two years) and 551 were temporary. Approximately 53% of these temporary drug switches were classified into group A (alimentary tract and metabolism), group C (cardiovascular system) and group N (nervous system) of the first Anatomical Therapeutic Chemical (ATC) level. 32 of the 551 shortages were categorized as expensive (add-on) (6%) and 156 were categorized specialty drugs (28%).
Conclusion
These results create an overview of the impact of drug shortages in a Dutch hospital. We saw a mixture of all therapeutic groups and a significant amount of specialty drugs. There was a strong increase in drug shortages in 2022 compared to 2021. Due to the increase in drug shortages and the impact on hospital pharmacies (workload, financial, and medication safety), further research is recommended. A national database for hospital pharmacies might be insightful and helpful.
Verantwoording
De hier opgenomen abstracts vormen de mondelinge presentaties tijdens de Nederlandse Ziekenhuisfarmaciedagen op 16 en 17 november 2023 te Arnhem.
Referentie
Citeer als: Nederlandse Ziekenhuisfarmaciedagen 2023. Nederlands Platform voor Farmaceutisch Onderzoek. 2023;8:a1774.
DOI
https://www.knmp.nl/resolveuid/529620d615484e90be0abbfd7b78610eOpen access

Reactie toevoegen