Nederlandse Ziekenhuisfarmaciedagen 2024
- Rubriek: Congresabstracts
- Identificatie: 2025;10:a1791
Donderdag 28 november - Sessie: Best abstract
Association of Busulfan Exposure and Outcomes after HCT for Patients with an Inborn Error of Immunity
Tim Bognàr a†*, Moises Garcia b†, Arief Lalmohamed ac, Tayfun Güngör d, Mathias Hauri-Hohl d, Susan Prockop e, Layne Oram e, Sung-Yun Pai ef, Jordan Brooks g, Rada M. Savic g, Christopher C. Dvorak g, Janel R. Long-Boyle g, Maja Krajinovic h, Henrique Bittencourt h, Anne-Charlotte C. Teyssier h, Yves Théoret h, Cary Martinez i, Toine C. G. Egberts ac, Erin Morales i, Mary Slatter j, Geoffrey D. E. Cuvelier k, Robert Chiesa l, Robert F. Wynn m, Mary Coussons m, Maria P. Cicalese nop, Marc Ansari qr, Susan E. Long s, Christen L. Ebens s, Hannah Lust t, Sonali Chaudhury t, Christa E. Nath u, Peter J. Shaw u, Steven J. Keogh u, M. Y. Eileen C. van der Stoep vwx, Robbert Bredius v, Caroline A. Lindemans yz, Jaap-Jan Boelens b† and Imke H. Bartelink a1a2†
a Department of Clinical Pharmacy, University Medical Center Utrecht/Wilhelmina Children's Hospital, Utrecht.
b Cell Transplantation and Cellular Therapies, Memorial Sloan-Kettering Cancer Center, New York, United States.
c Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
d Division of Stem Cell Transplantation and Children's Research Center, University Children's Hospital Zurich, University of Zürich, Zürich, Switzerland.
e Dana-Farber/Boston Children's Cancer and Blood Disorders Center, Boston, United States.
f Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health (NIH), United States.
g Departments of Allergy/Immunology/Bone Marrow Transplantation, Clinical Pharmacy, or Bioengineering & Therapeutic Sciences of the University of California San Francisco (UCSF), SanFrancisco, United States.
h Centre de Cancérologie Charles-Bruneau Centre de recherche - Hospital Sainte-Justine Montréal, Québec, Canada.
i Center for Cell and Gene Therapy, Baylor College of Medicine, Texas Children's Hospital, Texas, United States.
j Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle, United Kingdom
k Pediatric Blood and Marrow Transplantation, CancerCare Manitoba, University of Manitoba, Manitoba, Canada
l Great Ormond Street, Hospital for Children & Stem Cell Program, London, United Kingdom.
m Department of Blood and Marrow Transplant, Royal Manchester Children’s Hospital, Manchester, United Kingdom.
n San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), IRCCS San Raffaele Scientific Institute, Milan, Italy.
o Faculty of Medicine and Surgery, Vita-Salute S. Raffaele University, Milan, Italy.
p Pediatric Immunohematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy.
q Cansearch Research Platform in Pediatric Oncology and Hematology, Faculty of Medicine, Department of Pediatrics, Gynecology and Obstetrics, University of Geneva, Geneva, Switzerland.
r Division of Pediatric Oncology and Hematology, Department of Women, Child and Adolescent, University Geneva Hospitals, Geneva, Switzerland.
s Division of Pediatric Blood and Marrow Transplant & Cellular Therapy, MHealth Fairview Masonic Children's Hospital, University of Minnesota, Minneapolis, United States.
t Stem Cell Transplant Program, Ann & Robert Lurie Children's Hospital/Northwestern University, Chicago, United States.
u Children's Hospital at Westmead, Sydney, Australia.
v Department of Pediatrics, Willem Alexander Children's Hospital, Leiden University Medical Center.
w Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
x Center for Cell and Gene Therapy, Leiden University Medical Center.
y Department of Pediatrics, UMC Utrecht.
z Princess Máxima Center, Utrecht.
a1 Amsterdam University Medical Center, Location VUmc, Amsterdam.
a2 Cancer Center Amsterdam, Imaging and Biomarkers, Amsterdam.
† Authors have contributed equally.
* Correspondence: t.bognar-2@umcutrecht.nl.
Background
Previously, a clear relation between cumulative busulfan exposure (AUC) and clinical outcomes after pediatric and young adult allogeneic hematopoietic cell transplantation (HCT) was shown. As these studies involved only a small number of patients with inborn errors of immunity (IEI), the optimal target for this group remains unclear.
Objective
The objective of this study was to assess the optimal busulfan exposure in pediatric and young adult IEI patients.
Methods
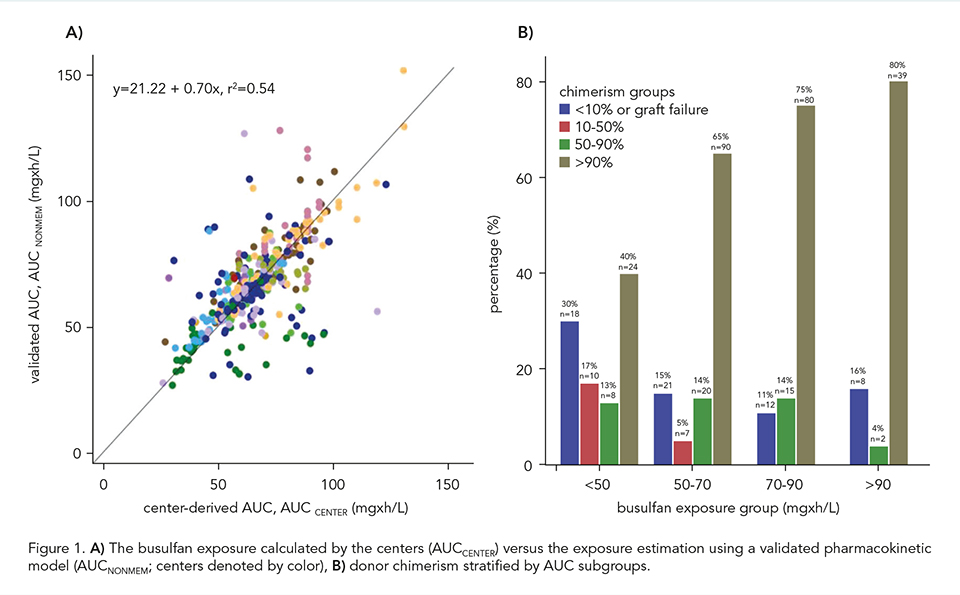
Patients who received a busulfan-based conditioning regimen between 2000 and 2023 from 17 centers were included. Our main outcome of interest was event-free survival (EFS); events considered were graft-failure (GF), and mortality. Other outcomes of interest were the most recent (myeloid or whole blood) donor chimerism. Patients were categorized into 4 IEI subgroups: combined immunodeficiency (CID), severe combined immunodeficiency (SCID), neutrophil disorders and hemophagocytic lymphohistiocytosis (HLH)-related disorders. Busulfan exposure was calculated by individual centers (AUCCENTER) and was re-estimated using all raw concentration-time profiles with nonlinear mixed effect modeling (AUCNONMEM) by applying an externally validated busulfan pharmacokinetic (PK) model (Bartelink, 2012). To assess the validity of the AUC prediction among centers, we compared the AUCCENTER with AUCNONMEM. To evaluate the busulfan AUCNONMEM in relation with the outcomes of interest, we used propensity score adjusted Weibull survival functions and Fine-Gray competing risk regression.
Results
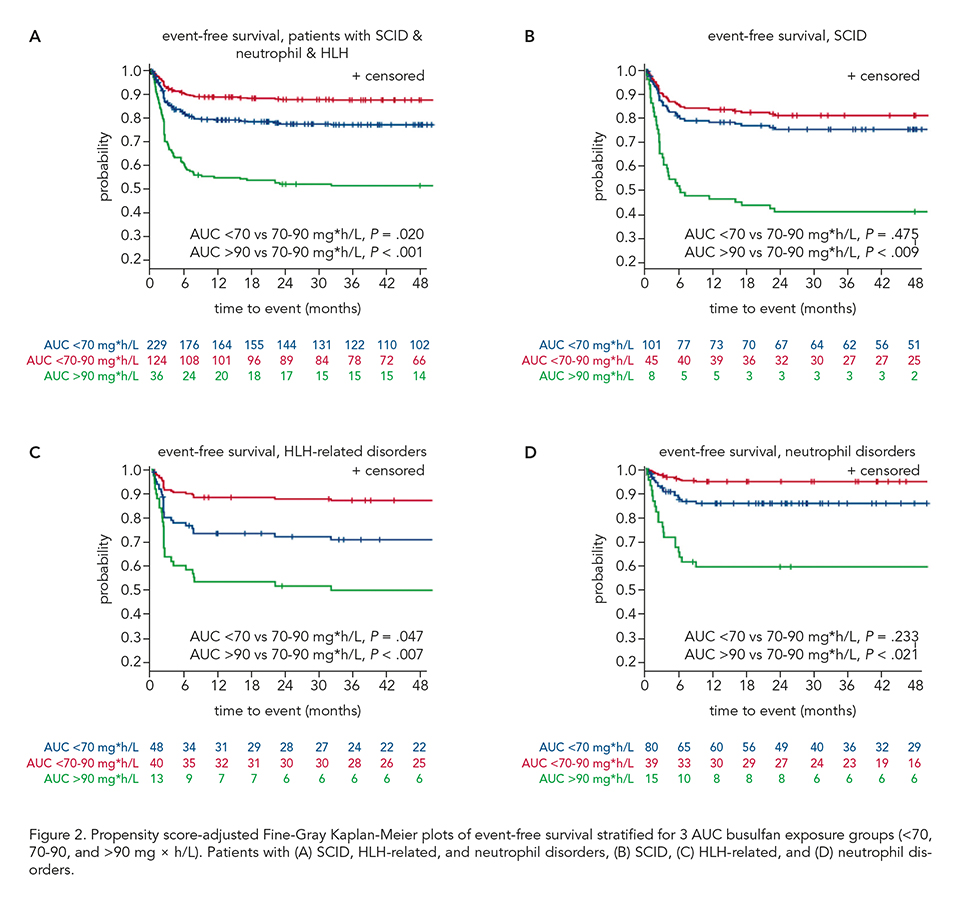
Overall, 562 patients were included: 154 (27.4%) SCID, 173 (30.8%) CID, 101 (18.0%) HLH and 134 (23.8%) neutrophil disorders. Median age was 1.7 years (range 0.08-27.0). CID disease subtype was an effect modifier (P = 0.03), therefore, patients with SCID, HLH-related, and neutrophil disorders were analyzed together (n = 389). The median busulfan AUCNONMEM was 69.0 mg×h/L and correlated poorly with AUCCENTER (r2 = 0.54; Figure 1A). In patients with the found optimal busulfan AUCNONMEM of 70-90 mg×h/L, 2-year EFS was 87.9% (95% confidence interval [CI] 80.3-92.6%), superior to < 70 mg×h/L (adjusted hazard ratio [HR] 1.97, 95% CI 1.11-3.49, P = 0.02), and > 90 mg×h/L (adjusted HR 5.05, 95% CI 2.43-10.49, P < 0.0001, Figure 2A-D). Donor chimerism increased with higher busulfan AUCNONMEM, plateauing at 90 mg×h/L (Figure 1B). For CID patients, an optimal AUCNONMEM for donor chimerism was found to be > 70 mg×h/L, while no optimal AUCNONMEM for EFS > 50 mg×h/L was found.
Conclusions
Improved EFS and higher donor chimerism may be achieved by targeting a cumulative busulfan AUCNONMEM of 80 mg×h/L (range 70-90). The data stresses the importance to uniformly use a validated population PK-model to estimate the AUCNONMEM.
De abstractpresentatie van Tim Bognàr werd bekroond met de prijs voor Best Abstract 2024.
Dose-Dependent Relationships in Prescribing Cascades: A Cohort Study
Ruveyda Gündogan-Yilmaz a*, Sadaf Wahedi a, Johanna H.M. Driessen bc, Atiya Mohammad ad, Petra Denig d and Fatma Karapinar abc
a Department of Clinical Pharmacy, OLVG, Amsterdam.
b Department of Clinical Pharmacy & Toxicology, Maastricht University Medical Center+.
c Department of Clinical Pharmacy, CARIM, Cardiovascular Research Institute Maastricht, Maastricht University.
d Department of Clinical Pharmacy and Pharmacology, University of Groningen, University Medical Centre Groningen.
* Correspondence: ruveyda_yilmazz@hotmail.com.
Background
Prescribing cascades occur when new so-called marker medications are prescribed to treat adverse drug reactions (ADRs) caused by an initial medication (index). This can lead to polypharmacy and increased healthcare costs. While dose reduction is often suggested as a strategy to mitigate prescribing cascades, the extent to which the dosage of an index medication affects the development of these cascades remains unknown.
Objective
This study aimed to investigate the dose-dependence of prescribing cascades across a range of medications.
Methods
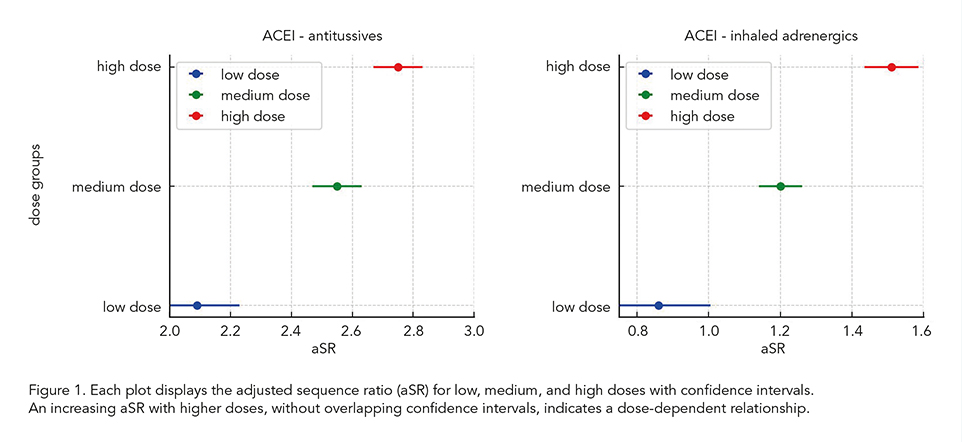
We performed a cohort study using prescription sequence symmetry analysis (PSSA) with data from over 600 Dutch community pharmacies. The relationship between different doses of index medications and the occurrence of 18 prescribing cascades was examined, including ACE inhibitors (ACEIs), statins, proton pump inhibitors (PPIs), diuretics, and antidepressants. Dose categories were determined using the World Health Organization (WHO) defined daily dose (DDD) classification, divided into low (< 0.50 DDD), medium (≥ 0.50 and ≤ 1.50 DDD), and high (> 1.50 DDD) dose groups. Adjusted sequence ratios (aSRs) were calculated, with an aSR greater than 1 indicating the occurrence of a prescribing cascade. A dose-dependent relationship was confirmed when aSRs increased with higher doses and their 95% confidence intervals (CIs) did not overlap.
Results
Of the 18 cascades analyzed, 12 showed a dose-dependent relationship. Notably, all seven ACEI-related cascades displayed a dose-dependent relationship. The aSR for ACEI-induced cough followed by antitussives increased from 2.09 (95% CI: 1.95-2.23) in the low-dose group to 2.75 (95% CI: 2.67-2.83) in the high-dose group. Similarly, for ACEI-induced cough followed by inhaled adrenergics, the aSR increased from 0.86 (95% CI: 0.71-1.00) in the low-dose group to 1.51 (95% CI: 1.44-1.59) in the high-dose group (Figure 1). Statins also exhibited dose-dependency in three of the six cascades. In contrast, no dose-response relationship was observed for cascades involving PPIs and diuretics.
Conclusions
These findings underscore the importance of dosage in managing prescribing cascades, particularly for ACEIs and possibly statins. Pharmacists and clinicians should remain vigilant for ADRs at higher doses and consider dose reduction as a strategy to reverse or prevent prescribing cascades. Further research is necessary to assess the effectiveness of dose adjustments in preventing ADRs and prescribing cascades.
Oral folinic acid prophylaxis prevents pemetrexed-induced neutropenia: results from a randomized clinical trial
R. Contrucci ab*, R. ter Heine b, M. Tonn a, J Diepstraten a, E van Thiel c, C van Kesteren d, M. van den Heuvel e, C. van der Leest f and N. de Rouw ab
a Department Clinical Pharmacy, Amphia Hospital, Breda.
b Department of Pharmacy, Radboud University Medical Center, Research Institute for Medical Innovation, Nijmegen.
c Department of Pulmonary Diseases, Albert Schweitzer Hospital, Dordrecht.
d Department Clinical Pharmacy, Albert Schweitzer Ziekenhuis, Dordrecht.
e Department of Pulmonary Diseases, Radboud University Medical Center, Nijmegen.
f Department of Pulmonary Diseases, Amphia Hospital, Breda.
* Correspondence: rcontrucci@amphia.nl.
Background
Pemetrexed is a cornerstone in the treatment of non-small cell lung cancer. Although this drug is generally well-tolerated, a substantial part of the patients receiving pemetrexed experience dose- or treatment-limiting toxicities, the foremost being neutropenia. Grade III/IV neutropenia has reported incidences up to 26% and can lead to hospitalization, treatment interruption, or even death. Based on in vitro and preclinical data pemetrexed-associated neutropenia can be prevented by treatment with prophylactic folinic acid.
Objective
The main objective of this study was to evaluate the effect of oral folinic acid in preventing pemetrexed-associated neutropenia.
Methods
A multicenter, open-label, double-arm, randomized trial was performed. Fifty patients treated with pemetrexed were randomized in a 1:1 ratio to either receive oral folinic acid 24 hours after pemetrexed administration for 3 days or receive standard of care without folinic acid. The primary endpoint was the difference in neutrophil count between both groups after the first cycle of chemotherapy at nadir. Secondary endpoints were the neutrophil count after the second cycle of chemotherapy, grade of neutropenia, efficacy of oncological treatment, renal function and the incidence of dose delays and reductions of pemetrexed.
Results
In total, 24 patients were included in the folinic acid group and 26 patients in the control group. Primarily, a higher absolute neutrophil count (P < 0.01) after the first cycle of chemotherapy was observed in the folinic acid group (median: 3.79; interquartile range [IQR]: 2.22-4.93) compared to the control group (median: 1.85; IQR: 1.43-3.78).
Secondarily, a higher neutrophil count (P = 0.01) was observed after the second cycle of chemotherapy in the folinic acid group (median: 2.60; IQR: 2.03-4.41) compared to the control group (median: 1.76; IQR: 0.87-2.73). The incidence of grade I neutropenia after the first cycle of chemotherapy was 4% in the folinic acid group vs. 27% in the control group (P = 0.04). The incidence of grade II neutropenia was 0% in the folinic acid group vs. 15% in the control group (P = 0.05). No differences were observed in the efficacy of treatment, renal function, dose reductions, delays, or discontinuation of treatment. Also no serious adverse events related to the treatment with folinic acid were observed.
Conclusions
Prophylaxis with oral folinic acid is effective in preventing pemetrexed-associated neutropenia and should be incorporated in the standard of care. Prospective evaluation after implementation may serve to validate our findings with respect to real-world reduction in toxicity and efficacy of lung cancer treatment.
Donderdag 28 november - Sessie 1: TDM en toxicologie
Lamotrigine use in lactating women: passage into breastmilk and infant exposure
D. den Besten-Bertholee a*, I. Wegner b, D.J. Touw cd, P.G.J. ter Horst a and P. Mian c
a Department of Clinical Pharmacy, Isala, Zwolle.
b Stichting Epilepsie Instellingen Nederland, Zwolle.
c Groningen Research Institute of Pharmacy, Section Pharmaceutical Analysis, University of Groningen.
d Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
* Correspondence: d.bertholee@isala.nl.
Background
Lamotrigine is an antiseizure medication used in the treatment of both epilepsy and bipolar disorder. Women of childbearing age are frequently prescribed lamotrigine, due to its relatively low teratogenic risk. It is known that lamotrigine use in lactation leads to detectable concentrations in breastmilk, with large interindividual variation.
Methods
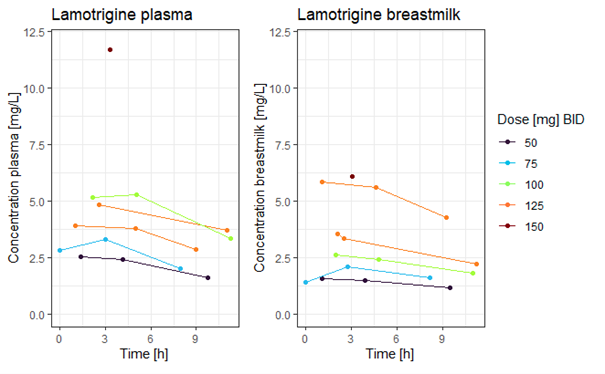
At our hospital we conducted an analysis of milk and plasma samples obtained from a limited cohort of lactating women (n = 6) using lamotrigine at dosages ranging from 50 to 300 mg per day, divided in two doses (n = 15 plasma and n = 16 milk samples). In the same cohort of lactating women using lamotrigine, we also measured single-point plasma concentrations in their suckling neonates (n = 5).
Results
The calculated Milk/Plasma ratio in our cohort, using an Area Under the Curve based approach, was 0.76 (range 0.47-1.2). The calculated Absolute Infant Dose was 0.29 mg/kg/day, which corresponds to a Relative Infant Dose of 10.1% (range 8.7-15.7%) in our cohort. The mean measured infant lamotrigine plasma concentration was 1.3 mg/L (range 0.5-2.2 mg/L).
Conclusions
Lamotrigine is excreted into breastmilk, and detectable levels are found in infants. However, the benefits of breastfeeding should not be overlooked. Higher blood concentrations generally lead to higher breastmilk concentrations, making it important to monitor blood concentrations in lactating women on lamotrigine.
Breastfed infants should be carefully monitored for side effects such as rash, drowsiness, poor weight gain or poor sucking.
Amanita poisonings: direct α-amanitin toxicity in the human kidney
K. Marais ab*, I.A.M. de Graaf b, D.J. Touw a and B.G.J. Dekkers a
a Department of Clinical Pharmacy and Pharmacology, University of Groningen, University Medical Center Groningen.
b Faculty of Science and Engineering, University of Groningen.
* Correspondence: f.s.marais@student.rug.nl.
Background
The deadly mushroom Amanita phalloides is responsible for the majority of fatal mushroom poisonings. As liver failure is the main cause of death in these poisonings, the effect of its primary toxin, α-amanitin, has been most closely studied on the liver. The results from these studies are the basis for the use of the antidotes benzylpenicillin and silibinin. Despite receiving these antidotes, 10-24% of patients still do not survive the poisoning. The role of the kidney in these poisonings remains largely unknown, but as many patients experience deterioration of kidney function in Amanita poisonings, understanding the interaction of the kidney with the amatoxins could be the key in improving outcome of patients.
Methods
Using human precision cut kidney slices (PCKS) we investigated the effects of α-amanitin (0.01- 5 µM) on cell viability (ATP assay), apoptosis (cleaved caspase-3), RNA expression (quantitative PCR) and α-amanitin uptake. Slices were exposed to α-amanitin for 4 and 48 hours.
Results
We demonstrate for the first time that α-amanitin is directly toxic to human kidney slices in a concentration dependent manner. The reduction in cell viability was associated with an increase in apoptosis and down-regulation of cMYC and MDM2, genes that promote cell proliferation. We also revealed that total RNA concentration decreases after 48 hours of exposure, which may be due to the primary mechanism of action of the toxin where RNA polymerase II is inhibited. These effects were associated with a concentration-dependent uptake of α-amanitin by the slices. Compared to liver, similar tissue concentrations were reached in kidney slices, but only after incubation with higher medium concentrations of α-amanitin. This corresponds to the degree of toxicity of α-amanitin in the kidney and liver slices, where liver toxicity occurs at lower medium concentrations of the toxin.
Conclusions
Our results demonstrate that α-amanitin may be directly toxic to the kidney. Given that acute kidney injury (AKI) can persist after apparent recovery from an Amanita poisoning, and that it may worsen liver damage, these results stress the importance of protecting the kidney along with the liver in Amanita poisonings. Further research into the mechanism of α-amanitin uptake into the kidney may reveal more effective targets for antidotes and improve outcomes.
The uptake mechanism of α-amanitin by hematopoietic cells
S.N. Botter a*, I.A.M. de Graaf b, W.F.J. Hof a, M. Visser a, D.J. Touw a and B.G.J. Dekkers a
a Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
b Faculty of Science and Engineering, University of Groningen.
* Correspondence: s.n.botter@student.rug.nl.
Background
Amanita phalloides, or death cap, is known for causing the most deaths due to mushroom poisoning. This is mainly due to the toxin α-amanitin, which may lead to acute liver failure. Uptake of the toxin in the liver is dependent on organic anion transporter polypeptide 1B3 (OATP1B3). Intracellularly, the toxin binds to RNA polymerase II, inhibiting protein synthesis which ultimately leads to cell death. Recently, we have shown that Amanita phalloides poisonings are also associated with hematotoxicity in patients. The uptake mechanism of α-amanitin by these cells is not known and may represent a new target in the treatment of α- Amanita phalloides poisonings.
Methods
Based on previous results, the HL60 cell line, a promyeloid cell line, was used as a model to study hematotoxicity. The effect of the toxin on cell survival in vitro was investigated by exposing cells to α-amanitin in the presence and absence of the antidotes silibinin and benzylpenicillin and the OATP1B3 inhibitors rifampicin and ciclosporin. Cell number was determined using an MTS assay. Intracellular α-amanitin was determined using an indirect competitive ELISA. HL60 cells were exposed to increasing concentrations of α-amanitin (0-10 µM) for different time periods (0-24 hours). In addition, whether uptake occurs by passive or active transport was examined by incubating cells at 4 and 37 °C.
Results
Results show that α-amanitin concentration-dependently reduced cell survival of HL60 cells after 72 hours of exposure. No effect on survival was observed during the first 24 hours of exposure. This effect of α-amanitin was not inhibited by the antidotes or OATP1B3 inhibitors. The decrease in cell survival was associated with a concentration- and time-dependent uptake of α-amanitin by the HL60 cells during the first 24 hours. This uptake was not observed when the cells were incubated at 4 °C, suggesting an active uptake mechanism.
Conclusions
Our results demonstrate that HL60 cells are sensitive to α-amanitin and can be a model for hematotoxicity in vivo. Uptake of α-amanitin by HL60 cells appears to be independent of the OATP1B3 transporter. However, uptake seems to take place via an active uptake mechanism. Identification of the mechanisms involved could lead to new interventions in Amanita phalloides poisonings.
Sessie 2: Klinische studies
Paracetamol Loading Dose Administration in Pediatric Population: A Retrospective Study
A.D.E. de Groot ab*, M.A.J. van der Wagt a, N.C. Punt ac, D.J. Touw ad, H.J. Verkade b and P. Mian a
a Department of Clinical Pharmacy and Pharmacology, University of Groningen, University Medical Center Groningen.
b Department of Pediatrics, University of Groningen, University Medical Center Groningen.
c Medimatics, Maastricht.
d Department of Pharmaceutical Analysis, Groningen Research Institute of Pharmacy, University of Groningen.
* Correspondence: a.d.e.de.groot@umcg.nl.
Background
Paracetamol (PCM) is one of the most frequent prescribed analgesic drugs in neonates and children worldwide. The varying amount of extracellular body water in children affects the volume of distribution (Vd) of paracetamol. In children, especially in preterm and term neonates, the increased Vd requires a higher initial dose of paracetamol per kilogram of body weight to achieve the same plasma concentrations as in adults. Accordingly, the Dutch Pediatric Formulary recommends a loading dose for (pre)term neonates and children with acute and postoperative pain. Although loading dose administration is clinically relevant, it has remained unclear if paracetamol loading doses are routinely administered in clinical practice.
Objective
This study evaluates the administration of paracetamol loading doses in an academic pediatric hospital and investigates the necessity of a paracetamol loading dose to achieve therapeutic target concentrations of 10 mg/L and steady state concentration (Css) in (pre)term neonates and children via pharmacokinetic simulations.
Methods
A retrospective study was performed including (preterm) neonates and children which were hospitalized between January 1st 2023 and January 1st 2024 and received at least one dose of intravenous or rectal paracetamol. The number of pharmacological treatments with paracetamol was evaluated, distinguishing between those in which a loading dose was administered and those in which a non-loading dose of paracetamol was administered ≤8 h prior to the start of the treatment. Pharmacokinetic simulations were performed using a previously developed population pharmacokinetic model to determine the effect of a loading dose on paracetamol Css.
Results
We included 911 intravenous and 1402 rectal treatment periods (1286 patients). A loading dose was administered in 21% and 1% of the cases, respectively. Paracetamol was administered ≤ 8 h prior in 42% and 52% of the intravenous and rectal treatment periods, respectively. Pharmacokinetic simulations shows that an intravenous or rectal loading dose will have peak plasma concentrations above Css and without a loading dose Css is reached after 12 h for intravenous and 18 h for rectal administration.
Conclusions
Our results indicate that in the vast majority of cases, paracetamol loading doses are not administered in an academic pediatric hospital. Loading doses were administered in only 21% and 1% at the start of paracetamol treatment. Supported by pharmacokinetic simulations, we conclude that the pharmacological treatment of (pre)term neonates and children with paracetamol can and should be improved, by administering a loading dose at the start of treatment.
Medication extravasation management and outcomes in patients who experienced an extravasation of medication during hospital admission: a mixed-method study
A.L. Bandringa a*, A. Keyany b, S. Saini b and B. Maat b
a Department of Pharmacoepidemiology and Clinical Pharmacology, Utrecht University.
b Department of Clinical Pharmacy, Elisabeth-TweeSteden Hospital, Tilburg.
* Correspondence: a.l.bandringa@students.uu.nl.
Background
Intravenous (IV) medication administration, a routine procedure in hospitalised patients, carries risks of complications such as extravasations. Extravasation, the unintentional leakage of a vesicant drug into surrounding tissue, can result in tissue damage and necessitates prompt intervention. Current literature lacks comprehensive data on the prevalence, management and patient outcomes associated with extravasations.
Objective
To determine the prevalence, nature and deployed management strategies of extravasations in hospitalised patients and to assess patient reported outcomes post-discharge.
Methods
A single-centre, retrospective, mixed-method study. All admitted patients who experienced extravasations between 01-01-2022 and 01-01-2024 were included. Eligible patients were invited to complete a questionnaire. Primary outcome: prevalence and nature of extravasations, including deployed management strategies. Secondary outcomes: patients’ outcomes after extravasation post- discharge, including their experiences regarding the extravasation during and after admission. Data were extracted from the Electronical Health Record (EHR) system and questionnaires. Data were analysed using descriptive statistics.
Results
We included 200 patients with 205 extravasations, yielding a prevalence of 0.014% (of IV administrations). Among the 74 different drugs involved, anti-infectives were most common (32.7%). Peripheral parenteral nutrition most frequently extravasated, both in absolute numbers (n = 19), as in relation to its number of administrations (4.2%), followed by acyclovir (n = 11 and 0.5% resp.). Extravasations predominantly occurred in the arm (61.5%) and were of moderate severity. All symptoms were most prevalent on the day of extravasation and mostly disappeared within 2-3 days. The top 3 symptoms were swelling n = 114 (70.8%), redness n = 66 (40.9%) and pain n = 62 (38.5%). Standard management strategies were applied in 96.6% of cases. Drug-specific management strategies particularly encompassed cold compresses alone n = 69 (33.7%) and hot compresses with hyaluronidase n = 65 (31.7%). The patient questionnaire (n = 41) revealed that 70.7% of patients mainly recalled experiencing swelling (70.7%), redness (48.8%) and pain (48.8%). A burning feeling was recollected far more often than it was documented in the EHR: n = 19 (46.3%) versus n = 1 (0.6%). When asked whether treatment of the extravasation had led to a reduction in symptoms, less than half of patients answered yes (41.5%). Post-discharge, 34.1% (n = 14) continued to suffer from symptoms.
Conclusions
This study is the first to comprehensively assess the prevalence, nature, deployed management strategies and patient outcomes after extravasation. The results indicate that there is room for improvement in management strategies and patient follow-up after extravasation.
Folinic acid prophylaxis enables safe chemo(immune)therapy with pemetrexed in lung cancer patients with renal impairment
Nikki de Rouw ab, Leila-Sophie Otten b, Mart P. Kicken c, Berber Piet b, Bonne Biesma d, Bianca van Veggel d, Christi M.J. Steendam c, Ben van den Borne c, Lizza E.L. Hendriks e, Sander Croes e, Anne-Marie C. Dingemans f, Daphne W. Dumoulin f, Ron H.J. Mathijssen f, Jacobus A. Burgers g, Alwin D.R. Huitema g, Hieronymus J. Derijks d, Maarten J. Deenen c, Michel M. van den Heuvel b and Rob ter Heine b*
a Amphia ziekenhuis, Breda.
b Radboudumc, Nijmegen.
c Catharina ziekenhuis, Eindhoven.
d Jeroen Bosch Ziekenhuis, ’s Hertogenbosch.
e MUMC+, Maastricht.
f Erasmus MC, Rotterdam.
g Netherlands Cancer Institute/Antoni van Leeuwenhoek, Amsterdam.
* Correspondence: r.terheine@radboudumc.nl.
Background
Pemetrexed is a cornerstone in chemo(immunotherapy) of non-small cell lung cancer and mesothelioma, however contraindicated in patients with renal impairment due to severe and potentially fatal toxicity concerns. Therefore, a large proportion of patients is withheld effective chemo(immunotherapy), that nonetheless have an indication for treatment. On a yearly basis, this concerns >100 patients in the Netherlands alone.
Objective
The aim of our study was to investigate the efficacy of oral folinic acid prophylaxis to prevent pemetrexed-induced toxicity in lung cancer patients with renal impairment and enable (chemo)immunotherapy in this vulnerable population.
Methods
We performed an intra-patient 3+3 dose escalation renal impairment study (eGFR < 45 mL/min). The pemetrexed dose was adjusted to renal function and patients received oral folinic acid prophylaxis 45mg four times daily on day 2-15 of each cycle. Study endpoints included safety (incidence of hematological and non- hematological toxicity, treatment delays) and pharmacokinetics compliant with regulatory guidances for renal impairment trials.
Results
Six patients with an estimated glomerular filtration rate (eGFR) between 26 and 41 mL/min were included. All patients were successfully escalated to the full dose. Adverse events patterns and pharmacokinetics were equivalent to those in patients with normal renal function. Grade I/II anemia occurred in five patients (already present at baseline). One occurrence of grade IV neutropenia was observed, which resolved without intervention. Moreover, in three patients a 1-week treatment delay occurred. Treatment resulted in response in 4 patients (n = 1 complete response, n = 2 partial response, n = 1 stable disease).
Conclusions
Pemetrexed can be safely administered in patients with impaired renal function when the dose is adjusted and folinic acid prophylaxis is administered, thereby enabling an effective treatment modality for patients that, thus far, could not be treated. As the study was compliant with regulatory guidance, the provided level of evidence in this study is sufficient to change clinical practice.
Impaired liver function: effect on paclitaxel toxicity, dose modifications and overall survival
Marieke Schmidt a, Robin Vernooij bcd, Merel van Nuland a, Erin Smeijsters a, Lot Devriese e, Nadia Haj Mohammad e, Thom Hermens a, Julian Stammers a, Christina Swart a, Toine Egberts af, Saskia Haitjema b and Laureen Lammers a*
a Department of Clinical Pharmacy, University Medical Center Utrecht.
b Central Diagnostic Laboratory, University Medical Center Utrecht, Utrecht University.
c Department of Nephrology and Hypertension, University Medical Center Utrecht.
d Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht.
e Department of Medical Oncology, University Medical Center Utrecht, Utrecht University.
* Correspondence: l.a.lammers@umcutrecht.nl.
Background
The anticancer drug paclitaxel is primarily metabolized in the liver. Previous studies have indicated a correlation between impaired liver function and paclitaxel toxicity, which may indicate dose reduction.
Objective
Since the evidence is limited, the aim of this study was to investigate the effect of impaired liver function on the hematological toxicity of paclitaxel, dose modifications and overall survival (OS).
Methods
For this single-center retrospective observational study, patients treated with paclitaxel for breast, esophageal and ovarian cancer at the University Medical Centre Utrecht between 2011 and 2022 were identified from the Utrecht Patient Oriented Database (UPOD). Based on regression analysis, the risk of developing Grade 3/4 hematological toxicity was compared between patients with normal and impaired (based on the NCI criteria for bilirubin and ASAT ( aspartate aminotransferase) concentrations) liver function. Additionally, differences in the occurrence of toxicity-related dose modifications and OS were evaluated between the two groups.
Results
A total of 569 patients were included. Breast cancer patients who were receiving advanced treatment and had mildly impaired liver function (ASAT ≤ 2x ULN, bilirubin ≤ ULN) had an increased risk of developing grade 3/4 neutropenia (HR = 4.39, 95% CI 1.20-16.02; P = 0.03). In addition, patients with impaired liver function treated according to the advanced ovarian cancer regimen had an increased risk of developing grade 3/4 leukopenia (HR = 12.64, 95% CI 2.12-75.22, P = 0.01) and dose modification (treatment discontinuation) (HR = 3.91, 95% CI 1.74-8.79, P < 0.01). Impaired liver function was also associated with decreased OS in inoperable esophageal and advanced ovarian cancer patients (HR = 7.65, 95% CI 2.54-23.1, P < 0.01 and HR = 2.98, 95% CI 1.36-6.54, P < 0.01, respectively). The risk of developing grade 3/4 hematological toxicity during lower-dose paclitaxel treatment protocols was not significantly different in patients with impaired liver function.
Conclusions
This study revealed that patients with impaired liver function treated with paclitaxel for breast and ovarian cancer in an advanced setting are at greater risk of developing hematological toxicity than patients with normal liver function at the start of therapy. Furthermore, in patients with ovarian (advanced) or inoperable esophageal cancer, impaired liver function is associated with decreased OS. Within these groups of patients, it is important to weigh the risk of upfront paclitaxel dose modifications versus an adaptive strategy.
The effect of interleukin-6 inhibitors as treatment for COVID-19 on PTSD, anxiety and depression 1 year after discharge in ICU-survivors
Thomas G. van Gelder a*, Irene J. van Diem-Zaal b, Arief Lalmohamed ac, Marieke Zegers d, Toine C.G. Egberts ac, Mark van den Boogaard d and Arjen J.C. Slooter e
a Department of Clinical Pharmacy, University Medical Center Utrecht, Utrecht University.
b Department of Intensive Care Medicine, Franciscus Gasthuis & Vlietland, Rotterdam.
c Division of Pharmacoepidemiology & Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University.
d Department of Intensive Care Medicine, Radboud University Medical Center, Nijmegen.
e Department of Intensive Care Medicine, University Medical Center Utrecht, Utrecht University.
* Correspondence: t.g.vangelder-2@umcutrecht.nl.
Background
Intensive care unit (ICU) survivors are at risk of developing mental health problems post- discharge. The potential of pharmacological interventions during ICU stays to prevent such symptoms remains underexplored. Research suggests that immune dysregulation, particularly elevated levels of interleukin-6 (IL-6), may contribute to the pathogenesis of mental health disorders such as depression. During the COVID-19 pandemic, the IL-6 inhibitors tocilizumab and sarilumab were incorporated into clinical guidelines for managing severe cases. Their use in the critically ill patient population presents a unique opportunity to investigate whether these agents can ameliorate post-ICU mental health problems, which are also present in COVID-19 ICU survivors specifically.
Objective
This study aimed to assess whether the use of IL-6 inhibitors during ICU treatment for COVID-19 is associated with reduced incidences of clinically significant post-traumatic stress disorder (PTSD), anxiety, and depression symptoms one year post-discharge.
Methods
This retrospective study used data from two prospective Dutch ICU follow-up cohorts: the University Medical Center Utrecht cohort (since 2011) and MONITOR-IC cohort (since 2017). Patients within these cohorts who were admitted to the ICU for COVID-19 and had one-year follow-up data by April 2023 were included. Mental health outcomes were assessed using validated questionnaires: the Impact of Event Scale for PTSD, the Hospital Anxiety and Depression Scale for anxiety and depression, and the EQ-5D for quality of life. Patients were categorized by IL-6 inhibitor (tocilizumab or sarilumab) exposure during or up to five days before ICU admission. The primary outcome was a composite measure of PTSD, anxiety, and depression symptoms one year post-discharge. Logistic regression analysis was used to evaluate the association between IL-6 inhibitor exposure and mental health outcomes, adjusting for confounders such as age, sex, BMI, APACHE-IV scores, and pre-ICU mental health status.
Results
A total of 288 COVID-19 ICU patients were included, with 145 (50%) receiving IL-6 inhibitors during admission. Mental health outcomes were available for 240 patients, of which 73 (30%) showed PTSD, anxiety, or depression symptoms. In the IL-6 inhibitor group, 35/122 (29%) developed these symptoms compared to 38/118 (32%) in the unexposed group. Logistic regression analysis showed an adjusted odds ratio of 0.74 (95% CI: 0.42–1.31), indicating no significant difference in mental health outcomes between the two groups.
Conclusions
In this retrospective study, we found no significant association between IL-6 inhibitor exposure during ICU admission and the development of PTSD, anxiety, or depression symptoms one year post-discharge.
Sessie 3: Real world studies
Palbociclib is safe for breast cancer patients with mild hepatic impairment: a multicenter retrospective study using real-world data
Alieke K. Bos a, Annelieke E.C.A.B. Willemsen b, Loes E. Visser c, Lennart J. Stoker d, Jurjen S. Kingma e, Mirjam K. Rommers f, Emile M. Kuck g, Paul D. van der Linden a and Merel van Nuland a*
a Department of Clinical Pharmacy, Tergooi Medisch Centrum, Hilversum.
b Department of Internal Medicine, Tergooi Medisch Centrum, Hilversum.
c Department of Clinical Pharmacy, Haga ziekenhuis, The Hague.
d Department of Clinical Pharmacy, Haaglanden Tergooi Medisch Centrum, The Hague.
e Department of Clinical Pharmacy, Ziekenhuisgroep Twente, Almelo.
f Department of Clinical Pharmacy, Noordwest Ziekenhuisgroep, Alkmaar.
g Department of Clinical Pharmacy, Diakonessenhuis, Utrecht.
* Correspondence: mvannuland@tergooi.nl.
Background
The liver is crucial for metabolizing the anticancer drug palbociclib, but limited information is available on the impact of hepatic impairment on its toxicity and efficacy, with no real-world data available.
Objective
This study aims to evaluate how hepatic impairment affects hematological toxicity and progression-free survival (PFS) of palbociclib in advanced hormone receptor- positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) breast cancer, using the National Cancer Institute scoring system, in a large real-world dataset.
Methods
This multicenter retrospective observational study included patients treated with palbociclib between August 2017 and February 2024. Regression analysis was used to compare the risk of developing grade 3/4 hematological toxicity between patients with normal and impaired liver function. Additionally, differences in PFS, dose reductions and liver toxicity were assessed.
Results
In total, 478 patients were included. Patients with hepatic impairment (n = 205) did not have an increased risk of developing grade 3/4 neutropenia compared to patients with normal hepatic function (n = 273) (RR = 1.10; 95% CI 0.83-1.46, P = 0.514). In addition, the PFS was not significantly different between both groups (HR = 1.14; 95% CI 0.92-1.42, P = 0.220).
Conclusions
In real-world settings, patients with mild hepatic impairment do not have a higher risk of developing palbociclib-induced neutropenia or disease progression than patients with normal hepatic function. These findings can guide clinicians when treating breast cancer patients with hepatic impairment.
Real-world experience with antenatal flecainide therapy: time to harmonize clinical practice?
J.A. Kroes ab*, M.G.P.J. Cox c, A.C.P. Wiesfeld c, F. Van den Heuvel d, W.Y. Vanagt d, S.J. Gordijn e, L.K. Duin e, D.J. Touw af and P. Mian a
a Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
b Department of Clinical Pharmacy and Pharmacology, Medical Centre Leeuwarden.
c University of Groningen, University Medical Center Groningen, Department of Cardiology.
d University of Groningen, University Medical Center Groningen, Department of Pediatric Cardiology.
e University of Groningen, University Medical Center Groningen, Department of Obstetrics and Gynaecology.
f University of Groningen, Groningen Research Institute of Pharmacy, Department of Pharmaceutical Analysis, Groningen.
* Correspondence: j.a.kroes@umcg.nl.
Background
Fetal arrhythmias, particularly supraventricular tachycardia (SVT) and fetal atrial flutter, are associated with severe complications during pregnancy. Flecainide is an antiarrhythmic used antenatally and aims to normalize fetal heart rhythm and prevent complications associated with persistent tachycardia.
Regularly, pregnant women are treated with flecainide for fetal arrythmias. However insight in the correct dose, whether the serum concentrations we obtain with these doses are within the target range (200-1000 mcg/L) and whether the dosages are adapted based on the measured concentrations is lacking.
Objective
The aim of the study was to retrospectively evaluate current practice regarding the antenatal treatment of fetal arrythmias, specifically with flecainide.
Methods
This was a retrospective, observational study. Using data from the lab-system GLIMS, we selected all women with a flecainide serum sample measurement between 1 January 2017 and 1 July 2024. Using electronic health record data, we selected women that were treated antenatally for fetal arrhythmias. We collected patient characteristics, flecainide concentrations, doses, reasons for dose adaptations, comedication, and timing of flecainide treatment in light of childbirth. We performed descriptive statistics using SPSS version 28.0.
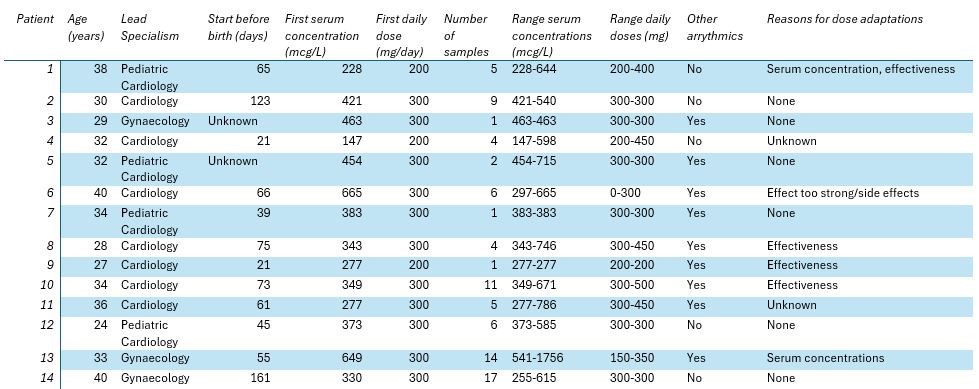
Results
Fourteen patients (aged 24-41) fulfilled our inclusion criteria (Table 1). In these patients, 86 serum levels were measured, ranging from 147-1756 mcg/L (target range 200-1000 mcg/L). The doses ranged from 200 to 450 mg per day. Eight patients were co-treated with other anti-arrhythmics (digoxin (n = 8) and sotalol (n = 1)). The number of samples varied from 1 to 17 per patient. Toxic serum levels (> 1000 mcg/L) were found in one patient and subtherapeutic (< 200 mcg/L) serum levels were found once in one patient. Reasons for dose adaptations were mainly based on effectiveness and in two cases based on flecainide levels.
Conclusions
We found wide variety in the number of samples analyzed per patient and the specialisms responsible for the patients. In addition, we found diversity in flecainide concentrations and associated doses. We currently do not know how to dose and when to measure flecainide and are confronted with a wide therapeutic range for flecainide. Knowledge regarding whether measuring flecainide improves fetal outcomes and the optimal therapeutic range would be a valuable addition to current practice. Patient characteristics and changing physiological and anatomical factors during pregnancy like renal function and enzyme expression might influence the flecainide concentration during pregnancy, but studies are lacking. In conclusion, our findings warrant harmonization of the current treatment strategies of women with fetal arrythmias, specifically for flecainide.
Identification of causes of drug shortages and mitigating interventions: a scoping review
N. Cornelissen ab*, S.W. Zielhuis a, P.M.L.A. van den Bemt c and B.J.F. van den Bemt bd
a Department of Pharmacy, Reinier de Graaf Gasthuis, Delft.
b Department of Pharmacy, Radboudumc, Nijmegen.
c Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen.
d Department of Pharmacy Sint Maartenkliniek, Nijmegen.
* Correspondence: nicky.cornelissen@rdgg.nl.
Background
Drug shortages form a significant global challenge that affect patient care and healthcare systems, leading to treatment delays, increased healthcare costs, and potentially severe health outcomes. Gaining more insight into the causes of drug shortages is crucial to develop interventions to mitigate their consequences.
Objective
This scoping review aims to provide an overview of the causes of drug shortages and the mitigating interventions in high-income countries.
Methods
This scoping review was conducted in accordance with the JBI methodology for scoping reviews. A comprehensive search was performed in four databases: MEDLINE (PubMed), Embase (Ovid), Web of Science and Cochrane Library, on December 21, 2023. Studies published in English or Dutch on the causes, management and mitigation of drug shortages in high-income countries were included. Two reviewers independently selected relevant studies via screening of title/abstract and full text. Relevant data on study characteristics, causes of drug shortages and interventions were extracted using a standardized form. The findings were analysed descriptively.
Results
The review included 39 studies out of 4,531 citations screened for eligibility. Causes of drug shortages (figure, upper part) included production issues, such as frequent manufacturing delays or termination, raw material shortages, and single supplier dependencies. Fluctuating demand and market forces leading to stock imbalances, poor supply chain management and regulatory issues, including batch release delays and recalls, strategic business decisions, pricing strategies, and external influences (natural disasters, cyber or terror attacks) are other causes or contributing factors to shortages. Several studies emphasized that the causes were frequently unknown. Interventions to manage drug shortages (figure, bottom part) included strategies such as procuring generic alternatives or different formulations, engaging alternative suppliers and wholesalers, and implementing measures like backordering, centralizing inventory, stockpiling, and extending shelf life to maintain adequate stock levels. Additionally, enhanced collaboration among pharmacists, as well as the use of import strategies, shortage alerts, and dashboards, were employed to improve management and response to shortages. While the causes appear to be associated with the early stages of the product life cycle, the interventions are mostly concentrated at the end (figure).
Conclusions
Currently, most interventions addressing drug shortages focus on temporary solutions at the end of the product supply chain, while the shortages originate earlier in the process. A focus on the causes early in the product chain is necessary for more sustainable solutions.

Monitoring multiple myeloma treatments in the Netherlands
J. Zwaveling a*, N.M.A. Blijlevens b, L.W. Tick c, H. ter Welle d and A. Sobels e
a Department of Clinical Pharmacy and Toxicology, Leiden University Medical Center.
b Department of Hematology, Radboudumc, Nijmegen.
c Department of Internal Medicine, Máxima Medical Centre, Eindhoven.
d PharmIntel, Ter Welle & Associates, Breukelen.
e Hospital Pharmacy, Haga Teaching Hospital, Den Haag.
* Correspondence: j.zwaveling@lumc.nl.
Background
Treatment effectiveness for real-world multiple myeloma (MM) patients vary greatly and multiple new medicines have been introduced over the last years, changing the treatment combinations and sequences of MM over time. Due to the introduction of these newly developed therapies, 5-year overall survival has doubled over the past decade to around 54%. However these new therapies come with high costs and at time of introduction the effectiveness is determined in a restricted population included in clinical trails only. Therefore, the NVvH and NVZA initiated the Dutch MM Treatment monitor in 2021 in order to gain insight in the effectiveness of MM treatments in clinical practice. The objective of our study was to assess the feasibility of construction a national monitor based on a real-world multiple myeloma population, focusing on use of real-world treatments.
Methods
An automated monitoring system is executed by PharmIntel and supervised by a board of hospital pharmacists and haematologists. From pseudonymised administrative healthcare data the following treatment related parameters are collected: medicine dispensing data and indication; patient parameters: age, gender, DOT and date of death. These data were used to calculate for each treatment: number of patients, treatment line, median duration, time-to-next-used treatment. Using digital algorithms, patient journeys and treatment sequences were constructed and were presented via a dynamic dashboard which allows correction for patient variables. Moreover, it is possible to benchmark outcomes between one's own hospital and other groups of hospitals.
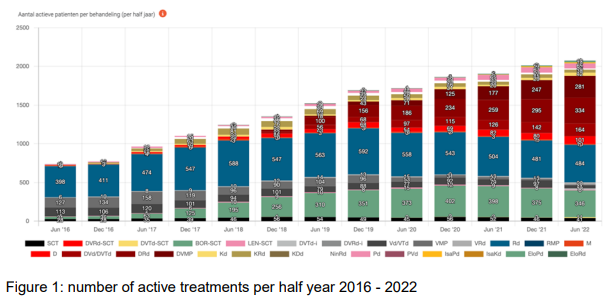
Results
In the period from 2012-2022, 50 Dutch hospitals participated in the MM monitor and uploaded data from in total 8477 patients. A cohort is created in which a total of 5761 patients started their first therapy after 2012, 2798 patients received a 2nd line and 1320 and 997 patients were treated with a 3rd and 4th line and further lines, respectively. Treatments changed significantly over time as shown in figure 1 and since the dashboard is dynamic, many analyses are possible such as median treatment duration per line or time to next used treatment, which is considered as a proxy for treatment free interval.
Conclusions
The Dutch MM Treatment Monitor is successfully constructed using real world data obtained from administrative healthcare data sources. Since the majority of the Dutch multiple myeloma patients is represented in this monitor, these data can be used to obtain insight of national use of newly introduced therapies in clinical Dutch practice.
Sessie 4: Procesoptimalisatie
Exploring Patient Perspectives on Drug Recalls: A Qualitative Analysis in the Netherlands
Pieter A. Annema ab*, Lenny M.W. Nahar-van Venrooij c, Marcel L. Bouvy d, Rob J. van Marum ab and Hieronymus J. Derijks a
a Department of Pharmacy and Clinical Pharmacology, Jeroen Bosch Hospital, ‘s-Hertogenbosch.
b Department of Elderly Care Medicine, Amsterdam Public Health Research Institute, Amsterdam UMC, Location VUmc, Amsterdam.
c Jeroen Bosch Academy Research, Jeroen Bosch Hospital, ‘s-Hertogenbosch.
d Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences (UIPS), Faculty of Science, Utrecht University.
* Correspondence: p.annema@jbz.nl.
Background
We previously demonstrated that drug recalls occur regularly in the Netherlands, with 44% of recalls requiring patients to change medication. The way drug recalls are communicated and handled may affect patients' trust in their medication and potentially affect their adherence to treatment. A reduction in medication adherence may lead to adverse health effects. Currently, it is unclear what the impact of a drug recall is on patients. To address this, we conducted a qualitative study to elucidate the experiences and preferences of patients concerning drug recalls in the Netherlands.
Methods
Two focus groups were conducted involving patients that experienced a drug recall, employing purposive sampling through an outpatient and a community pharmacy. The sessions were audio-recorded and transcribed verbatim. A thematic analysis approach was used for the analysis of the transcripts.
Results
Thirteen patients participated in the focus group sessions. Preliminary findings revealed that patients exhibited varying responses to drug recalls. Some patients displayed minimal concern and proceeded through the recall unaffected. Conversely, others reacted with heightened concern, prompting them to actively engage in the process. A common challenge was the lack of clarity in recall communication, as one patient noted: “Just be open and honest about it. And of course, in Jip-en-Janneke language, so that I understand it − I'm not a pharmacist”. Furthermore, patients expressed the need for more detailed information on what constitutes a contaminated drug, including its immediate and potential long-term effects. In terms of ways of communication, most patients emphasized the importance of timely, transparent communication through qualified personnel.
Conclusions
Preliminary conclusions indicate that healthcare providers must realize that patients may react differently to drug recalls and emphasize the importance of effective communication. Such communication should be accurate, clear, and easily understood, conveyed in a non-disturbing manner by the appropriate entity, delivered timely, and accompanied by opportunities for patients to seek clarification and ask questions. To further assess the implications for daily practice, we will conduct a questionnaire study with a larger population, aiming to support regulatory decision-making and improve the handling of drug recalls in the future.
Continued use of potentially inappropriate medication after hospital discharge: a retrospective cohort study
Judith de Ruijter-van Dalem ab*, Marjo Janssen c, Johanna HM Driessen ab, Carl E.H. Siegertd c, Alex Marmorale d, Daniala L. Weir d and Fatma Karapinar-Çarkit ab
a Department of Clinical Pharmacy & Toxicology, Maastricht University Medical Center+, Maastricht.
b Department of Clinical Pharmacy, CARIM, Cardiovascular Research Institute Maastricht, Maastricht University.
c Department of Clinical Pharmacy, OLVG Hospital, Amsterdam.
d Department of Internal Medicine, OLVG Hospital, Amsterdam.
e Epic Systems Corporation, Verona, Wisconsin, United States.
* Correspondence: judith.van.dalem@mumc.nl.
Background
Many patients receive temporarily indicated medications when they are hospitalized, e.g. opioids, benzodiazepines and/or antipsychotics. When such medications are continued at hospital discharge, it can result in potentially inappropriate medication (PIM) use post-discharge. However, there are no data available about continued use of PIMs at home in the Netherlands.
Objective
The aim of this retrospective cohort study was to investigate the incidence of continued use of in-hospital- initiated opioids, benzodiazepines, and antipsychotics post-discharge in the Netherlands.
Methods
A retrospective cohort study was conducted in a Dutch teaching hospital (OLVG). Patients aged ≥ 18 years and discharged from the hospital between Jan 2019-May 2023 with a new prescription of an opioid, benzodiazepine or antipsychotic started during hospitalization and continued at discharge, were included and followed-up for one year. Data from the hospital information system and dispensing records of community pharmacies (Nationwide Medication Record System) were used to identify patients who continued use post-discharge. Primary outcomes of this study were the incidence of continued PIMs prescriptions post-discharge for each of the medication classes of interest and duration of use. Duration of use was classified into < 30 days, 30-182 days and > 182 days. Secondary outcome was to investigate the most commonly prescribed individual medication of the PIM prescriptions. Descriptive statistics were used to analyze the data.
Results
Among 6,835 patients with a new prescription of one of the study medications, 82.7% were prescribed and dispensed an opioid at discharge (n = 5,652, mean age 61.1 years, 57.3% female), 14.7% a benzodiazepine (n=1,005, mean age 60.7 years, 53.3% female) and 2.6% an antipsychotic (n = 178, mean age 69.2 years, 48.9% female). 62.5% of new benzodiazepine users, 73.4% of new opioid users and 42.1% of new antipsychotic users had a duration of continued use of < 30 days post-discharge. A substantial number of patients had a duration of PIM use > 182 days post-discharge (13.4% of opioid, 20.9% of benzodiazepine and 36.0% of antipsychotic medication users). The most frequently initiated opioid was oxycodone (80.8%); for benzodiazepines this was oxazepam (41.3%) and temazepam (31.5%), and for antipsychotics this was quetiapine (39.3%) and haloperidol (34.8%).
Conclusions
This study found that antipsychotics were most often continued > 182 days post-discharge, followed by benzodiazepines and opioids. These results highlight the importance of recognizing PIMs and emphasize the need for careful evaluation of discontinuing these medications at hospital discharge or specifying a discontinuation date. Future work will assess indications for long-term use of these medications.
Discrepancy between home medication and medication at intensive care discharge in patients surviving a period of severe acute illness
F.N. Polderman a, H.J. Derijks a*, M.A. Sikma b and R.J. van Marum ac
a Jeroen Bosch Hospital, ‘s-Hertogenbosch.
b University Medical Center, University Utrecht.
c University Medical Center, Amsterdam.
* Correspondence: j.derijks@jbz.nl.
Background
During a period of acute illness requiring intensive care unit (ICU) admission, a substantial portion of home medication is discontinued. Part of this stopped medication needs to be reintroduced before ICU discharge. If indicated and not reintroduced, this could have significant consequences.
Objective
The aim of this study was to gain insight into the discrepancy between home medication and medication at ICU discharge.
Methods
In this retrospective cohort study, the medical record of patients admitted to the ICU of a non-university teaching hospital between January 2020 and September 2022 were studied. We aimed for inclusion of 200 patients. Inclusion criteria: ICU admission ≥ 48 hours, and alive at discharge from the ICU (without palliative care), and with registered home medication at hospital admission. Exclusion criteria: previous ICU stay during same hospital admission, transfer from and to another hospital and patients on chronic ventilation with an elective ICU admission. Medication was classified according to ATC-classification.
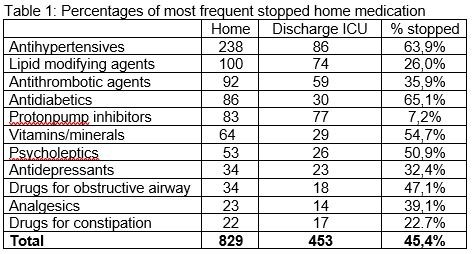
Results
We included 200 patients (mean age 63.5 years, 63.0% male, surgical admission 24.0%). 49.0% of patients were ventilated and 52.0% had no additional nutrition. Mean APACHE4 score was 68.4 and mean maximum SOFA score was 5.4. Median length of ICU stay was 93.3 hours (IQR 69.4-141.7). Mean number of home medication on admission and at discharge ICU were 5.0 and 2.7, respectively. Table 1 shows percentages of most frequent stopped home medication. Of all stopped home medication at ICU discharge, 22.4% was incorrectly not reintroduced.
Conclusions
Almost half of used home medication was stopped during a period of acute illness requiring ICU admission. For several home medication, failure to reintroduce could lead to significant consequences for patients and should be subject of future research.
The effect of a pharmaceutical discharge letter on unintentional continuation beyond the stop date for complex anticoagulation
Renate van Uden ab, Marcia Vervloet c, Patricia Pols d, Karina Meijer b, Patricia van den Bemt b and Matthijs Becker a*
a Spaarne Gasthuis, Haarlem/Hoofddorp.
b University Medical Center Groningen.
c Nivel, Netherlands Institute for Health Services Research, Utrecht.
d Heemsteedse Apotheek, Heemstede.
* Correspondence: mbecker@sahz.nl.
Background
Guidelines advise to use combined antithrombotic therapy for a limited time period, varying from one week to one year. Combined antithrombotic therapy is usually started during hospital admission and the intended stop date is often after discharge. Adequate information transfer to the community pharmacy is important, in order to prevent unintentional continuation.
Objective
We aimed to examine the effect of a pharmaceutical discharge letter sent to the community pharmacy on unintentional continuation after the intended stop date of combined antithrombotic therapy.
Methods
This intervention study with a pre-post design was conducted in the Spaarne Gasthuis hospital. Patients discharged with combined antithrombotic therapy were eligible for inclusion. For patients included in the pre-intervention group (May 2018 - August 2019) and post-intervention group (August 2020 - May 2021), the antithrombotic therapy was reviewed during the admission by a hospital pharmacist (resident) for guideline compliance, as part of usual care. For the patients in the post-intervention group, a pharmaceutical discharge letter was sent to their community pharmacy, with information about the antithrombotic therapy, the indication and the intended stop date. The primary endpoint was the proportion of antithrombotic therapies that were dispensed after the intended stop date, based on dispensing data from the LSP and the PHARMO database. Data were analysed using a Chi2 test.
Results
We could evaluate 227 of 442 patients (51.4%) in the pre-intervention period and 371 of 645 patients (57.5%) in the post-intervention period. Reasons for not being evaluable were missing dispensing information (26.6%), changes in therapy or new events (7.8%) and death or premature discontinuation (10.7%). For 38 patients on triple therapy, two stop dates were analysed. For patients with an intended duration of less than one month, 6 of 123 patients (4.9%) had antithrombotic medication dispensed after the intended stop date in the pre-intervention period versus 4 of 196 patients (2.0%) in the post-intervention period (P = 0.16). For patients with an intended duration of more than one month, these numbers were 51 of 135 patients (37.8%) versus 31 of 182 patients (17.0%), respectively (P < 0.001). Fifteen of 51 patients (29.4%) and 8 of 31 patients (25.8%) had antithrombotic medication dispensed more than 180 days after the intended stop date.
Conclusions
A pharmaceutical discharge letter reduces the risk of unintentional continuation after the intended stop date, in patients with an intended duration of more than one month. For every five letters sent, one unintentional continuation after the intended stop date is avoided.
Medication reviews during hospitalisation: impact on the number of medications and the medication use complexity in older patients with polypharmacy
C.M. Falke a*, F. Karapinar bc, M. Bouvy a, W. Knol d and A.C.G. Egberts ae
a Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University.
b Department of Clinical Pharmacy & Toxicology, Maastricht University Medical Center+.
c Department of Clinical Pharmacy, CARIM Cardiovascular Research Institute Maastricht, Maastricht University.
d Geriatric Medicine Department and Expertise Centre Pharmacotherapy in Old Persons, University Medical Centre Utrecht.
e Department of Clinical Pharmacy, University Medical Centre Utrecht.
* Correspondence: c.m.falke@uu.nl.
Background
Medication reviews with (older) patients reduce medication-related problems, but studies have shown inconsistent improvements in clinical outcomes. In medication reviews, explicit criteria such as STOPP/START-criteria are often used to optimise medication. However, these criteria pay little attention to the medication use complexity. Addressing medication use complexity is important as this is associated with lower quality of life, more (re)hospitalisations, and lower medication adherence.
Objective
This study aimed to evaluate the effect of medication reviews during hospitalisation on the number of medications and medication use complexity in older patients with polypharmacy.
Methods
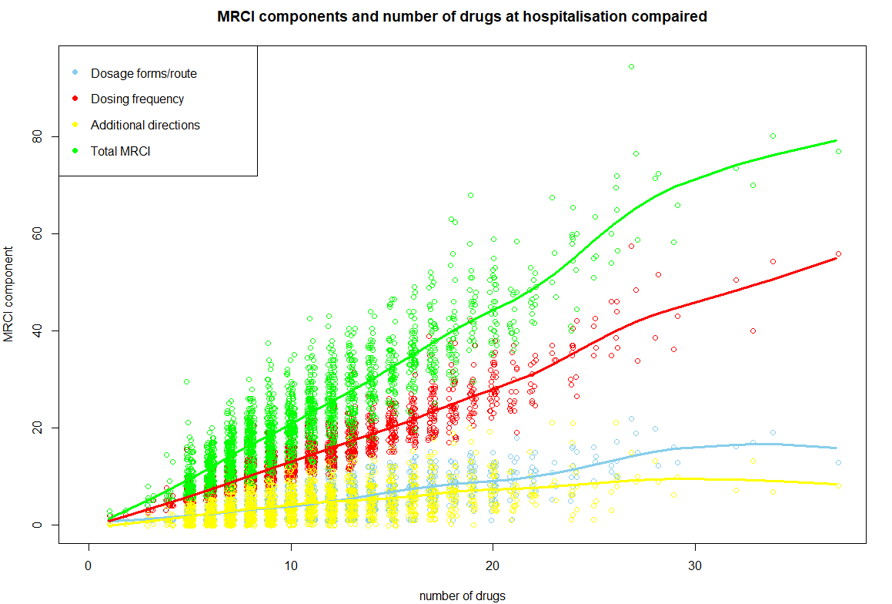
This follow-up study used data from the cluster-randomised OPERAM trial conducted in four European countries (the Netherlands, Belgium, Switzerland, and Ireland). Hospitalised patients aged ≥ 70 years, with ≥ 3 chronic conditions and ≥ 5 chronic medications were included. In the intervention group, patients received a medication review from a trained pharmacotherapeutic team (consisting of a geriatrician and pharmacist), supported by software using the STOPP/START-criteria. The control group received standard care. The outcome of this follow-up study was the difference in the number of chronic medications and medication use complexity between hospital discharge and admission, comparing the intervention and control group. Medication use complexity was measured using the Medication Regimen Complexity Index (MRCI), which assesses three domains: dosage route/form, dosing frequency, and additional directions concerning administration (e.g. medication intake on an empty stomach). Descriptive analyses, correlation graphs on medication use complexity and its domains versus the number of medications, and Chi-square tests were performed.
Results
A total of 1,923 patients were analysed, with a mean age of 79.4 (SD 6,3) years, 45% were female, and the mean number of medications used was 10,5 (SD 4,5). A strong correlation (r = 0,91) was found between the number of medications and the medication use complexity. In the intervention group, patients were discharged with an average of 0.8 (SD 3.8) more medications, and medication use complexity increased by an average of 3.1 points (SD 9.7). In the control group, patients were discharged with an average of 1.2 (SD 3.3) more medications, and medication use complexity increased by an average of 3.6 (SD 8.8) points.
Conclusions
During hospitalisations, the number of medications and medication use complexity both tend to increase. A medication review focused on the STOPP/START criteria has limited impact on this increase. Medication use complexity should be considered as an additional factor in medication reviews.
Sessie 5: Procesoptimalisatie
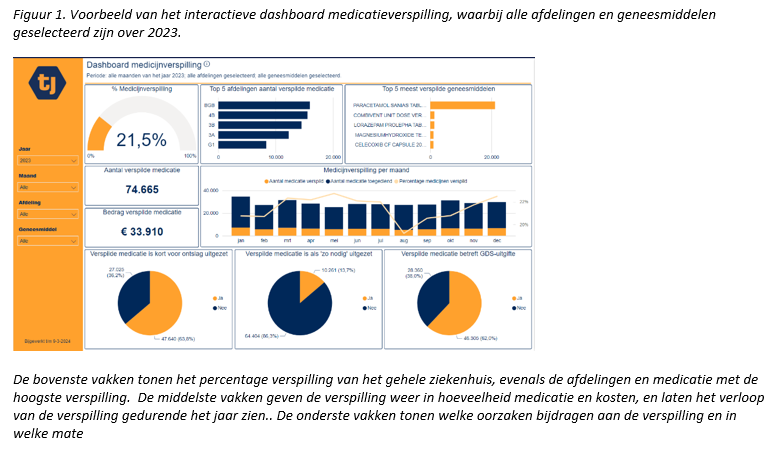
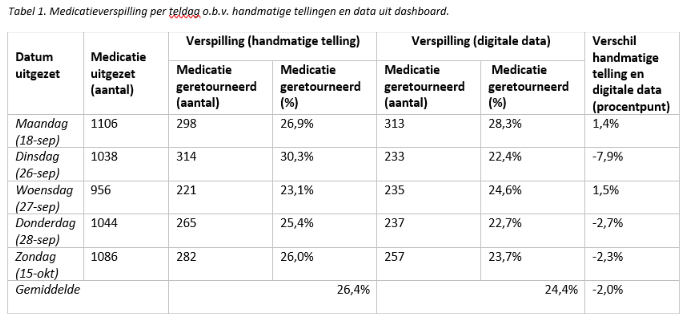
Making healthcare more sustainable: development and validation of a medication waste dashboard
C. Jonker, M. Huisman, J. Grassi, D. Mitrovic and M. Jongsma *
Tjongerschans Ziekenhuis, Heerenveen.
* Correspondence: minke.jongsma@tjongerschans.nl.
Background
Healthcare contributes to climate change, making sustainability in healthcare an important objective. This has resulted in the participation of hospitals in the Green Deal 3.0 ‘sustainable care’ in order to reduce the ecological footprint of healthcare.
Objective
Developing and validating a dashboard to provide insight into different aspects of medication waste in hospitals.
Methods
A prospective observational validation study. Medication dispensed but not administered (and hence returned to the hospital pharmacy) was defined as waste. This was both counted manually for five days, as well as generated digitally by using corresponding digital prescription data. To validate the dashboard, the medication waste percentages from the manual counts were compared with the digitally generated data.
Results
No significant difference was found between the medication waste percentages based on manual counting and the digitally generated data (P-value 0.24).
Conclusion
The validation of the dashboard has been successful, demonstrating that it can be used reliably to gain insight into medication waste in a hospital.
Enhancing Parenteral Medication Safety by using Safety II: Only double-check if it truly adds value
A. van de Plas *, D. Klein, F. Karapinar-Çarkit and R. Rennenberg
Maastricht UMC+.
* Correspondence: a.vande.plas@mumc.nl.
Background
In order to prevent medication errors, according to the Dutch guideline, all parenteral medication administrations have to be double-checked. In daily practice, the compliance of double- checking is insufficient while medication errors still occur. Research from 2019 by Schutijser et al. showed that nurses conduct their own risk-assessment to decide whether or not to use a double- check. Furthermore, double-checking requires a considerable investment of valuable nursing time. Our hypothesis is that if double-checks are solely applied in high-risk administrations, where they do have added value, overall medication safety will improve.
Objective
In this study the effect of risk-based double- checking on safe administration performance in parenteral medication administration is measured.
Methods
In a controlled before-and-after trial on two comparable nursing wards (intervention and control ward), the effects of risk-based double-checking on the safe administration performance were studied through disguised observation.
Using Failure Mode Effect Analysis (FMEA), all parenteral medication administrations were classified into high and low-risk categories by a multidisciplinary team consisting of pharmacists, nurses, and clinicians. For low-risk administrations, barcode verification was used. For high-risk administrations, in addition to barcode verification, a second nurse must double-check the administration route and administration rate, which are not verified by barcode. The risk-based policy was implemented bottom-up.
Administrations were classified as performed safely when all six items (patient, medication, dose, administration time, administration route, and administration rate) were double-checked by barcode verification and/or a second nurse.
Results
After implementing the risk-based double-check, safe performance of parenteral medication administration increased from 0% (0/90) to 41% (22/54) on the intervention ward, saving 48 minutes of nursing time per day. Post-intervention, in 83% (5/6) of high-risk administrations, a double-check was applied on the intervention ward compared to 29% (2/7) of high-risk administrations on the control ward.
Conclusions
Our results indicate that risk-based double-checking enhances safety in parenteral medication administration compared to double-checking all administrations, and also saves nursing time. Our recommendation is to apply the double-check only in high-risk administrations where this additional control measure truly adds value. Further studies are required to prove the effect of this strategy on medication errors and the associated risk of harm.
This project exemplifies a best practice of Safety II, where safety is enhanced by addressing the gap between work-as-imagined and work-as-done.
Characteristics of hospital pharmacy residents
R.J. Zaal a*, F. van Rosse a, N. Vink-van Kimmenade b, N. Veldhorst-Janssen c, E.M. Kemper d, P. van der Linden e, S. Kabadayi f, K.M. Stegers-Jager b and I. Wilting g
a Erasmus MC, University Medical Center Rotterdam.
b Radboud University Medical Center, Nijmegen.
c Maastricht University Medical Centre.
d Apotheek A15, Gorinchem.
e Tergooi MC, Hilversum.
f Franciscus Gasthuis & Vlietland, Rotterdam.
g University Medical Center Utrecht.
* Correspondence: r.zaal@erasmusmc.nl.
Background
In our multicultural society, with 14% of inhabitants with a non-western migration background, a diverse healthcare workforce is needed to represent and serve the patient population. Besides, creating equal career opportunities for pharmacists with different backgrounds is essential form a social justice point. For optimal pharmaceutical care, it is also important to obtain a balance between hospital pharmacists specialized in patient care and those specialized in product care.
Objective
The objective of this study was to gain insight into the extent of diversity amongst hospital pharmacy residents, in terms of gender, ethnic background and interest in patient care versus product care, and into the criteria being used in selection procedures.
Methods
A semi-quantitative cross-sectional study using electronic questionnaires among hospital pharmacy residents and supervisors (i.e. hospital pharmacists responsible for the residency program in their hospital) was conducted. Residents who started their residency between 2018 and 2023 were eligible for participation and were asked for gender, ethnic background and chosen differentiation. Supervisors were asked for the criteria used in the selection of residents and the importance of these criteria (scale 1 to 5).
Results
In total, 131 of 169 (78%) invited residents and 34 of 42 (81%) supervisors gave consent for participation. Of the participating residents 30% were male and 18% had a non-western migration background. The gender distribution and ethnic diversity in their team were said to taken into account during the selection procedure by 72% and 68% of the supervisors, respectively. For both gender and ethnic diversity their median scored importance was 3 (range 1-5). A differentiation primarily focused on patient care (clinical pharmacology or a specific pharmacotherapeutic area) was chosen by 61% of the residents; 12% of the residents primarily focused on product care (manufacturing & pharmaceutical analysis, radiopharmacy or nuclear medicine). The balance between a priori interest in patient care versus product care was taken into account during selection by 57% of the supervisors (importance 3.5, range 2-5).
Conclusions
Our results are a starting point to improve awareness for creating a diverse population of hospital pharmacists, reflecting our multicultural society and meeting the future needs for our workforce. Future research should focus on selection procedures and potential loss of diversity in the transition from pharmacy student to resident. Besides, in the transition from resident to hospital pharmacist, the balance between availability and needs for hospital pharmacists with specific differentiations should be monitored.
Reducing medication waste in nursing facilities through interventions based on a baseline measurement: a pre-post implementation study
Minou van Seyen a*, Anniek Middeldorp a, Elise A.J. Smulders b, Mark Niestijl a, Pieter Annema a, Elisabeth A.M. Laro c, Charlotte L. Bekker d and Hieronymus J. Derijks a
a Department of Pharmacy, Jeroen Bosch Hospital, 's-Hertogenbosch.
b Utrecht University, School of Pharmacy, Utrecht.
c Van Neynsel, 's-Hertogenbosch.
d Department of Pharmacy, Radboud Institute for Health Sciences, Radboud University Medical Centre, Nijmegen.
* Correspondence: m.v.seyen@jbz.nl.
Background
The increasing aging population in the Netherlands is driving a higher demand for nursing care. Concurrently, there is a growing focus on sustainable medication management within nursing facilities. This is driven by the environmental impacts associated with medication waste, as well as the escalating medication shortages and financial costs. At present, there is a lack of comprehensive data regarding the magnitude of medication waste within nursing facilities and the effectiveness of potential strategies to mitigate this issue.
Objective
This study aims to quantify the extent of medication waste and develop and evaluate interventions to reduce this medication waste.
Methods
In this prospective pre-post intervention study data on medication waste for the eldercare institution Van Neynsel was collected for a four-week period each. In total 416 beds with departments for somatic patients, psychogeriatric patients, first-line care, and geriatric revalidation were included. Wasted medication was defined as unused medication being disposed of, quantified in units, weight, and value (euro). Nursing staff in representative departments recorded reasons for wastage using a standardized form. During the implementation phase interventions were developed and prioritized based on the waste assessment and focus groups with healthcare professionals (i.e. nurses, physicians, pharmacy assistant, pharmacist).
Results
Here we present the preliminary results of the pre-intervention measurement and intervention development. The total amount of medication waste over four weeks was 12,163 units, 41 kilograms, and had a value of € 5,462. Departments with shorter lengths of stay, first-line care and geriatric revalidation, exhibited higher levels (5-fold) of medication waste.
Paracetamol was the most frequently wasted medication, 2,236 units (18%) overall. Laxatives represented a high-value group, with 867 units weighing 14.2 kilograms (34%). Inhalation medication, with 85 units, had a value of € 1,188 (22%).
Key targets for intervention included enhancing awareness among healthcare providers, optimizing inventory management, improving medication transfer between primary care and facilities, refining quantity of dispensed medication and the medication ordering process. Proposed interventions encompassed the inclusion of macrogol and paracetamol in the departmental inventory, reducing the supply of as-needed and multi-dose medications, and increasing the involvement of nursing staff in the process.
Conclusions
This study identified substantial medication waste in terms of quantity, weight, and costs, revealing opportunities for waste reduction. Interventions targeting the full medication process were designed for waste minimization. During the post-intervention measurement, the effectiveness of the implemented measures will be determined.
Vrijdag 29 november - Sessie 1: Bereidingen
Implementation of a 3D printer in a hospital setting
Eveline E.M. van Kampen a*, Javier E. Maduro a, Khair Aljaja a, Anne Schäfer-van Raalte b, Hugo Hoogeveen a, Anton P. Aulbers c, Renz J. van Ee c, Thomas A. Smits b, Henk Spijker b and Liesbeth J. Ruijgrok a
a Department of Hospital Pharmacy, Erasmus MC, University Medical Center Rotterdam.
b Ziekenhuisapotheek, Maasstad Ziekenhuis, Rotterdam.
c The Netherlands Organization for Applied Scientific Research (TNO), Eindhoven.
* Correspondence: e.e.m.vankampen@erasmusmc.nl.
Background
The necessity for individualized dosages has led to the exploration of 3D-printing technology for producing personalized medicines. This technology allows dosage, size, shape, and taste adjustments based on individual patient characteristics. Our ongoing joint project with TNO at Erasmus MC and Maasstad Ziekenhuis aims to implement the semi-solid extrusion (SSE) print technique in clinical practices to produce personalized medicines.
Objective
In this study, we aim to describe all necessary steps to implement this innovative technology in the daily care routine of a hospital.
Methods
An inventory of facility requirements for 3D pharma printing was made. A 3D pharma- printer was previously developed by TNO. We jointly developed User Requirements Specifications (URS), Standard Operating Procedures (SOP) and Factory Acceptance Test (FAT) protocol. End-users performed the FAT at TNO. At Erasmus MC, we evaluated if the 3D printing process is workable and user-friendly. After a training session, pharmacy personal printed 3D placebo tablets independently. This approach ensured participants were well-trained and confident in using the printer while providing a comprehensive evaluation of the printer’s performance under different conditions. Following up on the outcomes of the first part of the study, we developed Installation Qualification (IQ), Operational Qualification (OQ), Performance Qualification (PQ) and Process validation protocols. These protocols ensure that the 3D printer meets the technical and functional requirements. Finally, the qualification and validation part will be performed at Maasstad Ziekenhuis, where the printer will be made operational for clinical use.
Results
The described requirements of premises for 3D pharma printing can be used to assess implementation of 3D printing technique in a hospital setting. The FAT was performed successfully, and no deviations were found. The user test significantly improved the Standard Operating Procedure (SOP) including preparation procedures, printer use, and software. The SSE 3D printed tablets showed high precision and consistency, meeting the quality requirements uniformity of mass and weight distribution outlined in the European Pharmacopoeia (EP) and Laboratory of Dutch Pharmacists (LNA) standards. The qualification and validation activities based on IQ, OQ, PQ will be performed by October 2024.
Conclusions
This work resulted in the availability of relevant documents and practical experience about the implementation, qualification and validation of 3D pharma printing in a hospital setting. These results can support when defining the strategy of hospitals to start using this innovative technique.
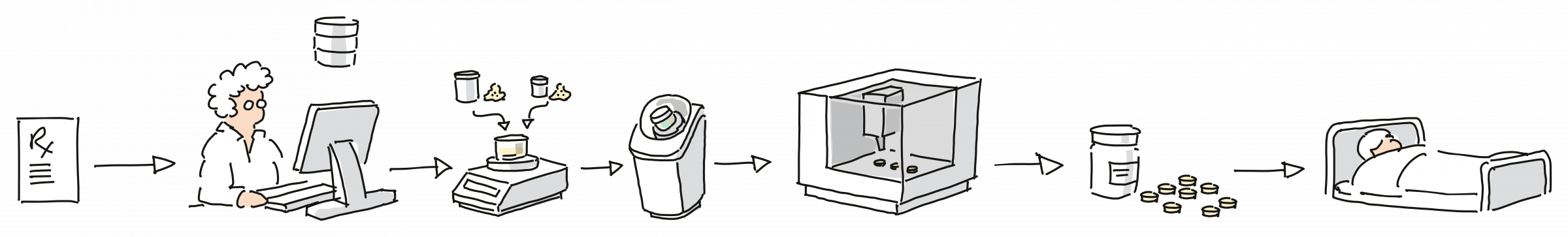
Accuracy testing of aseptic compounding using the semi-automated SmartCompounder Chemo robot
T. Mulders *, D. Maas, B. Nuijen and B.A.W. Jacobs
Department of Pharmacy and Pharmacology, The Netherlands Cancer Institute-Antoni van Leeuwenhoek, Amsterdam.
* Correspondence: t.mulders@nki.nl.
Background
Compounding of intravenous medications in hospital pharmacies is a critical process that demands high accuracy to ensure patient safety. Semi-automated compounding systems among which SmartCompounder Chemo robot (Smartcompounders, Enschede, The Netherlands) have been introduced to support aseptic compounding.
Objective
This study aimed to determine the accuracy of the SmartCompounder robot in our hospital pharmacy setting.
Methods
Accuracy testing of the SmartCompounder Chemo robot was conducted for the following three scenarios of admixture preparation using 50 mL saline bags: 1) single vial of 24 mL dummy concentrate 2) two 10 mL and one 4 mL vial of dummy concentrate and 3) two 24 mL vials of dummy concentrate. For each run, 10 saline bags were filled. All three testing scenarios were performed in triplicate. Accuracy criteria were based on the following criteria: 98% of preparations have a maximum deviation of 5% relatively to the nominal weight and the bias in gravimetric control is < 1%.
Results
The SmartCompounder Chemo robot demonstrated acceptable performance for compounding of admixtures. Average (range) deviations from the nominal dummy dose were −0.85% (−1.1% - −0.7%), -1.17% (−0.95% - −1.41%) and −0.68% (−0.92% - −0.5%) for scenario 1, 2 and 3, respectively. Overall, the average bias in gravimetric control was 0.24%. The highest observed bias in gravimetric control was 0.51%. All scenarios met validation criteria, with 98% of preparations deviating < 5% from nominal weight and bias in gravimetric control < 1%.
Conclusions
The SmartCompounder Chemo robot meets the stringent accuracy requirements for the preparation of admixtures using IV bags. Deviations in nominal filling weight were low and gravimetric control using the SmartCompounder Chemo robot is precise. Future validation work includes sterility testing. Furthermore, the performance of semi-automated filling of Folfusors will be studied. Current findings support the adoption of compounding robots as a valuable tool in aseptic compounding.
An efficient and flexible system for the prescription and preparation of parenteral nutrition admixtures for pediatric oncology patients
R. Lange a*, J.J.M. Hassink a, N.C. van der Linden-de Munk b, J. Dekker b and L.M. Hanff a
a Pharmacy, Princess Máxima Center for pediatric oncology, Utrecht.
b Dietetics, Princess Máxima Center for pediatric oncology, Utrecht.
* Correspondence: r.lange-2@prinsesmaximacentrum.nl.
Background
Princess Máxima Center is the largest pediatric oncology center in Europe. Some patients require parenteral nutrition (PN) due to disease related malnutrition and/or treatment related gastrointestinal toxicity. PN is preferably combined with (minimal) enteral nutrition (EN) when possible.
For the preparation of PN-admixtures three compartment bags are used with standard additions of micronutrients and extra macronutrients and/or electrolytes when needed.
Objective
In this study the system used for the prescription and preparation of PN-admixtures in our center and the nutritional intake (PN and EN) in comparison to nutritional needs were evaluated.
Methods
We developed standardized medication orders for PN, a spreadsheet for physicians to choose the best fitting source bag allowing for comparison of prescribed amounts of nutrients with ESPGHAN guidelines and a spreadsheet for pharmacists to calculate the amount of macronutrients and/or electrolytes to be added to the bag(s), taking into account the infusion rate.
The PN-bags prepared from 2021 to 2023 and the nutritional intake (PN and EN) of 50 random PN- patients were retrospectively assessed and compared with their energy and amino acid/protein requirements calculated by the dietitians. If the administered total of macronutrients (both energy and amino acids/protein) in the PN and EN was at least 80% of the initial calculation, the intake was considered adequate.
Results
In the study period 329 children received PN, for a median period of 16 days (range: 1-475 days). Numeta® (G16E, G19E) or Olimel® (N5E, N7E, N4E, N9) were used as the source bag. In total, 10109 PN-bags were prepared, of which 26% were lipid free, 12% contained extra additions and 3% were electrolyte restricted. 98.5% of the PN-bags were administered through a central venous line. In all cases, standard three compartment bags could be used as the source. The main reasons for modification of the standard PN composition were fluid restriction, need for lipid reduction and electrolyte disturbances.
Based on an evaluation of the nutritional data of 50 random PN-patients, we concluded that the nutritional requirements were met in 94% of the cases.
Conclusions
We developed an efficient and flexible system for the prescription and preparation of PN- admixtures for pediatric oncology patients, based on the use of three compartment bags and self- developed spreadsheets for physicians, dietitians and pharmacists.
By simply adjusting the standard composition, nutritional requirements of the patients in our center can almost always be met with these PN-admixtures, in combination with EN.
Photosensitivity of furosemide in parenteral preparations
R. Lange a and A. van Loon b
a Pharmacy, Princess Máxima Center for pediatric oncology, Utrecht.
b Laboratory of Dutch Pharmacists (LNA), The Hague.
* Correspondence: r.lange-2@prinsesmaximacentrum.nl.
Background and objective
Princess Máxima Center is the largest pediatric oncology center in Europe. To ensure optimal product quality of parenteral preparations, almost all parenterals are prepared ready to administer (RTA) in our pharmacy. Due to the large number of preparations, there is a great need to organize the preparation process as efficiently as possible.
The preparation process of the light-sensitive furosemide is inefficient. After the preparation process, the syringes are individually packed in a light-proof bag. This requires double labelling and extra handling during product release.
In order to make this process more efficient, we investigated the influence of light on the decomposition of furosemide during the entire process from preparation to administration to the patient.
Methods
The concentration of furosemide injection fluid prepared in our center ranges from 0.5 mg/mL (diluted with sodium chloride 0.9%) to 10 mg/mL (undiluted).
The only relevant exposure to light occurs during administration, which takes place within 5 minutes for bolus injection, within 30 minutes if administered with a pump or (in rare cases) continuously. Only LED lighting is present throughout our center.
For the illumination experiments, furosemide solutions were prepared gravimetrically in three concentrations: 0.5 mg/mL, 2.5 mg/mL and 10 mg/mL (undiluted). Twelve syringes were prepared for each concentration. The syringes were illuminated for 8 h, 24 h, 72 h and 168 h, respectively, with LED light of 800 lux. Each exposure experiment was performed in triplicate. Because it was known that photodegradation can lead to discoloration, all samples were visually assessed after exposure.
All samples were analysed by HPLC using the method for related substances of the British Pharmacopoeia (BP). Because no impurities were found, the concentration of each sample was determined with a UV measurement.
Results
After exposure to light, all solutions were clear and colorless.
For all 36 samples, none of the impurities exceeded the detection limit of 0.02%, except for impurity D, which was ≤ 0,03%. BP rejects all results smaller than 0.05%. No significant decrease in concentration was found in any of the samples with different exposure time to LED light.
Conclusions
In furosemide injection fluid no impurities, no discoloration and no decrease in concentration were found after exposure to LED light of 800 lux for up to 168 h. Therefore it is concluded that under the conditions in our center no measurable photodegradation occurs in these products. We will therefore no longer protect the products against light, which makes the process more efficient.
Sessie 2: TDM en toxicologie
Quantification of Ivacaftor, Tezacaftor, Elexacaftor, and Lumacaftor and Their Active Metabolites in Plasma Using UHPLC-MS/MS: Doors Open to the Application of Therapeutic Drug Monitoring in Cystic Fibrosis Treatment
A.M.A. Wessels a, F.A. Elzinga a*, G.H. Koppelman b, O.W. Akkerman c, P. Mian a and D.J. Touw a
a Afdeling Klinische Farmacie en Farmacologie, Universitair Medisch Centrum Groningen.
b Afdeling Kinderlongziekten en Kinderallergologie, Beatrix Kinderziekenhuis, Universitair Medisch Centrum Groningen.
c Afdeling Longziekten en Tuberculose, Universitair Medisch Centrum Groningen.
* Correspondence: f.a.elzinga@umcg.nl.
Background
An ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) method was developed to quantify the cystic fibrosis transmembrane conductance regulator (CFTR) modulators, ivacaftor, tezacaftor, elexacaftor, and lumacaftor and their active metabolites hydroxymethyl ivacaftor, tezacaftor M1, and N-desmethylelexacaftor in human EDTA plasma.
Methods
The analytical method utilized protein precipitation with stable isotope dilution for sample preparation, facilitating a simple and rapid assay, with a total runtime of only 2.1 minutes. Separation of the seven components and stable isotope-labeled internal standards was achieved on a C18 column, followed by detection using a tandem quadrupole mass spectrometer. Validation of the method was conducted in accordance with the ‘Bioanalytical Method Validation Guidance for Industry’ of the Food and Drug Administration and with European Medicines Agency’s ‘Guidance on bioanalytical method validation’.
Results
The assay covers concentrations ranging from 0.010-10 mg/L for ivacaftor, hydroxymethyl ivacaftor and N-desmethylelexacaftor, from 0.025-25 mg/L for elexacaftor and tezacaftor, from 0.050-50 mg/L for tezacaftor M1 and from 0.100-100 mg/L for lumacaftor, using a sample volume of 10 µL. Matrix comparison confirmed the applicability of the assay to human serum and heparin plasma. Stability experiments indicated stability of the CFTR modulators in EDTA plasma over ten days under different conditions. At room temperature, all seven components remained stable for eight days and for ten days in the refrigerator in EDTA plasma and in EDTA whole blood. All seven components were stable in EDTA plasma for ten days in the autosampler after sample preparation and through four freeze-thaw cycles.
Conclusions
An accurate UHPLC-MS/MS method with a simple sample preparation has been developed and validated for the quantification of elexacaftor, tezacaftor, ivacaftor and their metabolites. The developed assay was applied to investigate exposure to these compounds in a CF patient who did not experience therapeutic benefits from Kaftrio.
A new ELISA-based assay for the quantifcation of α-amanitin in biological matrices
Bart Dekkers a*, Erik Jacobs a, Dimitra Gkolfi a, Kim Kleefman a, Nomie Botter a, Kik Marais a, Daan Touw a and Inge de Graaf b
a Department of Clinical Pharmacy and Pharmacology, University of Groningen, University Medical Center Groningen.
b School of Science and Engineering, University of Groningen.
* Correspondence: b.g.j.dekkers@umcg.nl.
Background and objective
Amanita phalloides, also known as the death cap, is one of the deadliest mushrooms known to man. Mortality rates may rise up to 40% in patients not treated with antidotes. Amanita mushrooms contain several toxins of which α-amanitin is considered to be the main toxin that cause systemic effects. Progress in the field is currently hampered by the lack of a specific method for the quantification of α-amanitin in biological matrices. Moreover, development of a method could improve treatment of poisoned patients.
Methods
An enzyme-linked immunosorbent assay (ELISA) was developed to quantify α-amanitin in tissue, cellular, blood and urine samples using a monoclonal antibody specific for α-amanitin (AMA9C12, Bever et al, toxins, 2018).
Results
A competitive ELISA was developed using increasing α-amanitin concentrations to select the optimal coating concentration. Blocking of the plate was performed with bovine serum albumin. Incubations with increasing concentrations of antibody and the antigen were performed in a separate non-absorbing plate. After washing of the coated plate, antigen/antibody mixtures were added. Bound antibody was visualized by incubation with the secondary antibody followed by tetramethylbenzidine (TMB) incubation. Formation of tetramethylbenzidine diimine (blue) was stopped by addition of sulphuric acid. Absorbance was measured at 450 nm. After optimization, a method was established with a lower limit of quantification (LLoQ) of ~2,5 ng/mL and an upper limit of quantification of ~25 ng/mL. A small, but variable matrix effect was observed for the various matrices tested. This effect could be corrected by normalization of the absorbances to those measured in the control without antigen. Preliminary data demonstrate that the method can be used to measure cellular α-amanitin concentrations in liver and kidney tissue slices and in in vitro cell samples exposed to the toxin. In addition, the ELISA can quantify α-amanitin in spiked urine samples and in urine of patients with an Amanita phalloides poisoning.
Conclusions
We developed a new ELISA-based method for the quantification of α-amanitin in various biological matrices. This method can be used to better study the toxicokinetics of the toxin, both on a patient as wells as cellular level.
Pharmacokinetic data of lopinavir/ritonavir in second-line treatment of African children living with HIV
Anne Kamphuis a*, Hylke Waalewijn ab, Alexander Szucbert c, Chishala Chabala d, Mutsa Bwakura- Dangarembizi e, Shafic Makumbi f, Joan Nangiya g, Vivian Mumbiro e, Veronica Mulenga d, Victor Mussiime g, David Burger a, Diana Gibb c, Angela Colbers a and THE CHAPAS4 TRIAL TEAM
a Department of Pharmacy, Radboud Research institute for Medical Innovation (RIMI), Radboudumc, Nijmegen.
b Division of Clinical Pharmacology, Department of Medicine, University of Cape Town, South Africa.
c Medical Research Council Clinical Trials Unit at University College London, United Kingdom.
d University Teaching Hospital, Lusaka, Zambia.
e University of Zimbabwe Clinical Research Centre, Harare, Zimbabwe.
f Joint Clinical Research Centre, Mbarara, Uganda.
g Joint Clinical Research Centre, Kampala, Uganda.
* Correspondence: anne.kamphuis@radboudumc.nl.
Background and objective
Lopinavir/ritonavir (LPV/r) is widely used in second-line for children living with HIV. As pediatric 100/25mg tablets have inconsistent availability in some countries, using adult 200/50 mg tablets could simplify procurement. For children weighing 25-34.9 kg, the daily dose of 600/150 mg could be achieved by dosing two 200/50mg tablets in the morning and one in the evening, instead of three 100/25 mg tablets twice-daily (current WHO-recommendation). In CHAPAS-4, second-line treatment options for children with HIV were investigated. We performed a PK substudy to evaluate LPV/r exposure in children with different nucleoside reverse transcriptase inhibitor (NRTI) backbones.
Methods
Children with HIV aged 3-15 years failing NNRTI-based first-line treatment were randomized to 4 anchor drugs with emtricitabine/tenofovir alafenamide or standard-of-care NRTIs. Children weighing 14–24.9 kg received 200/50 mg LPV/r twice-daily; 25–34.9 kg received 400/100 mg LPV/r in the morning and 200/50mg in the evening; ≥ 35 kg received 400/100 mg LPV/r twice-daily. At steady-state, PK samples were taken (pre-dose, 1, 2, 4, 6, 8, 12h) after LPV/r intake. LPV concentrations were measured using a HPLC method. Lopinavir AUC0-12h, Cmax, and Ctrough were calculated using non- compartmental analysis. PK data of children receiving similar dosage were used for comparison.
A one-way ANOVA on log-transformed values was performed to assess differences in LPV exposure between weight-bands and backbones.
Results
51 children were included in this substudy; 11 were excluded. There were 9 children in the 14-19.9 kg weight band, 10 in 20-24.9 kg, 9 in 25-34.9 kg and 12 in ≥ 35 kg. The overall Geometric Mean (GM), (CV%) AUC0-12h was 116.15h*mg/L (37%) and the GM (CV%) Cmax was 12.52 mg/L (32%). The GM (CV%) Ctrough of 7.71 mg/L (52%) in this sub-study is ∼57>trough associated with dyslipidemia in adults (> 8 mg/L). The Ctrough and Cmax were significantly higher in children weighing 25-34.9 kg (11.7 and 15.38 mg/L) compared to children weighing 14-19.9 kg and ≥ 35kg (6.19 and 12.52 mg/L; 6.96 and 12.10 mg/L, respectively). These children received a higher milligram per kilogram body weight dose in the morning compared to the other weight-bands. There were no differences in LPV exposure when comparing the backbones.
Conclusions
This data provides evidence for adequate exposure of LPV/r in children 3-15 years of age weighing ≥ 14 kg in second-line treatment when using adult tablets down to 25 kg, however limitations of toxicity and pill burden should be considered.
Silibinin and benzylpenicillin as antidotes in human precision-cut liver slices exposed to α-amanitin
Kim Kleefman ab, Bart Dekkers a*, Daan Touw a and Inge de Graaf b
a Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen, University of Groningen.
b Faculty of Science and Engineering, University of Groningen.
* Correspondence: b.g.j.dekkers@umcg.nl.
Background and objective
Foraging of mushrooms is growing in popularity. While most mushrooms are edible, about 3% are toxic. The death cap mushroom (Amanita phalloides) is among the deadliest. This variety contains α-amanitin, a toxin that targets the liver and binds to RNA polymerase II, ultimately leading to liver failure. Despite the use of antidotes like silibinin and benzylpenicillin, treatment remains inadequate due to limited understanding of the mechanism of action of both the toxin and the antidotes.
Methods
This study examined the effects of α-amanitin on human precision-cut liver slices (hPCLS) and the protective properties of silibinin and benzylpenicillin during 4- and 48-hour incubation periods. We focused on the viability of the hPCLS using an ATP assay. Intracellular concentrations of α- amanitin were determined by cELISA and gene expression of MDM-2 and c-MYC were analyzed by qPCR. Given their short half-lives, c-MYC and MDM-2 mRNA could serve as markers for the impact of α-amanitin on RNA polymerase II activity.
Results
After 48 hours of exposure, both silibinin and benzylpenicillin prevented α-amanitin-induced reduction in viability of the hPCLS considerably. Moreover, silibinin appeared to be more effective at higher toxin concentrations, providing significantly better protection.
The cELISA results showed that the concentration of α-amanitin in the hPCLS increased linearly with the medium concentration. The uptake was partially reduced by the antidotes, although the effects were not statistically significant. Moreover, the results suggest that after 4 hours of exposure, a steady state in α-amanitin uptake was already reached, with toxin concentrations remaining constant. After 4 hours of incubation, a decrease in c-MYC and MDM-2 expression was observed in the hPCLS exposed to α-amanitin and silibinin appeared to reduce this effect, although these findings were not statistically significant. This effect could be due to decreased mRNA production. After 48 hours, MDM-2 expression decreased at higher doses of α-amanitin and silibinin increased the expression, while c- MYC was elevated in the groups treated with the toxin only. This suggests possible compensatory mechanisms after prolonged toxin exposure.
Conclusions
hPCLS appear to be a suitable model for studying liver toxicity in Amanita poisoning, as a dose-dependent toxicity is evident in the slices. Both silibinin and benzylpenicillin work as antidotes during α-amanitin exposure by improving viability and possibly reducing toxin uptake, with silibinin showing the greatest effect. c-MYC and MDM-2 appear not be reliable markers for α-amanitin toxicity, possibly due to their involvement in apoptosis. Further research is needed to confirm these findings.
Sessie 3: Klinische studies
Short-course aminoglycosides as adjunctive treatment in adults with sepsis: a cluster randomized trial
Sharon E.J.D. van den Eijnde ab*, Eva L. Koekenbier b, Paul D. van der Linden ab, Jan Jelrik Oosterheert c, Marc J.M. Bonten b and Cornelis H. van Werkhoven b on behalf of the SAGA study team
a Department of Pharmacy, Tergooi Medical Centre, Hilversum.
b Julius Centre for Health Sciences and Primary Care, University Medical Centre Utrecht.
c Department of Internal Medicine, University Medical Centre Utrecht.
* Correspondence: svandeneijnde@tergooi.nl.
Background
Cephalosporin monotherapy or cephalosporin-aminoglycoside combination therapy are both used as first-choice empirical antibiotic treatment for sepsis in the Netherlands as there is no convincing evidence favouring one of both options.
Objective
We hypothesized that ceftriaxone or cefuroxime monotherapy (‘monotherapy’) is non-inferior to cefuroxime combined with short-course treatment with aminoglycosides (gentamicin or tobramycin, ‘combination therapy’) for the empirical treatment of community-acquired sepsis.
Methods
A cluster-randomized cross-over trial was conducted in nine Dutch hospitals, which were randomized to monotherapy or combination therapy. Adult patients with a severe community- acquired sepsis, of suspected urinary, abdominal, or unknown origin, were eligible. The primary endpoint was 30-day all-cause mortality, with a non-inferiority margin defined as an adjusted absolute risk difference (aARD) of less than 5%. Secondary endpoints included hospital re-admission, and nephrotoxicity using the Kidney Disease Improving Global Outcomes criteria. Regression analyses were conducted using multiple imputed data and propensity score overlap weighting.
Results
The study was stopped prematurely prior to the first cross-over for slow overall recruitment and 46% non-compliance to protocol in the combination therapy group. Between May 2022 (start first hospital) and May 2023, 451 patients were included: 158 treated with combination therapy and 293 with monotherapy. Urinary tract infections were the most prevalent focus of infection, followed by unknown focus of infection (Table 1). The overall median NEWS at admission was 7.00 (IQR 6.00-9.00) and 41.9% of the patients had one or more treatment restrictions at admission. 30-day mortality was 11.4% (18 patients) in the combination therapy group versus 17.1% (50 patients) in the monotherapy group, yielding an adjusted odds ratio of 0.81 (95% CI 0.38-1.75) and an aARD of −2.7% (95% CI −13.9-7.0; point estimates in favour of monotherapy). Statistically significant differences between treatment groups were not observed for any of the secondary outcomes (Table 2). Of 393 patients who survived the initial hospitalization, 37 (9.4%) were re-admitted within 30 days after- discharge, with no difference between treatment groups. Additionally, there were no differences in the incidence of nephrotoxicity between patients treated with combination therapy or monotherapy.
Conclusions
There is no indication that combination therapy improves clinical outcomes, although the study was insufficiently powered to demonstrate non-inferiority of monotherapy. The data suggest that the treatment regimens are equally safe with regards to nephrotoxicity.

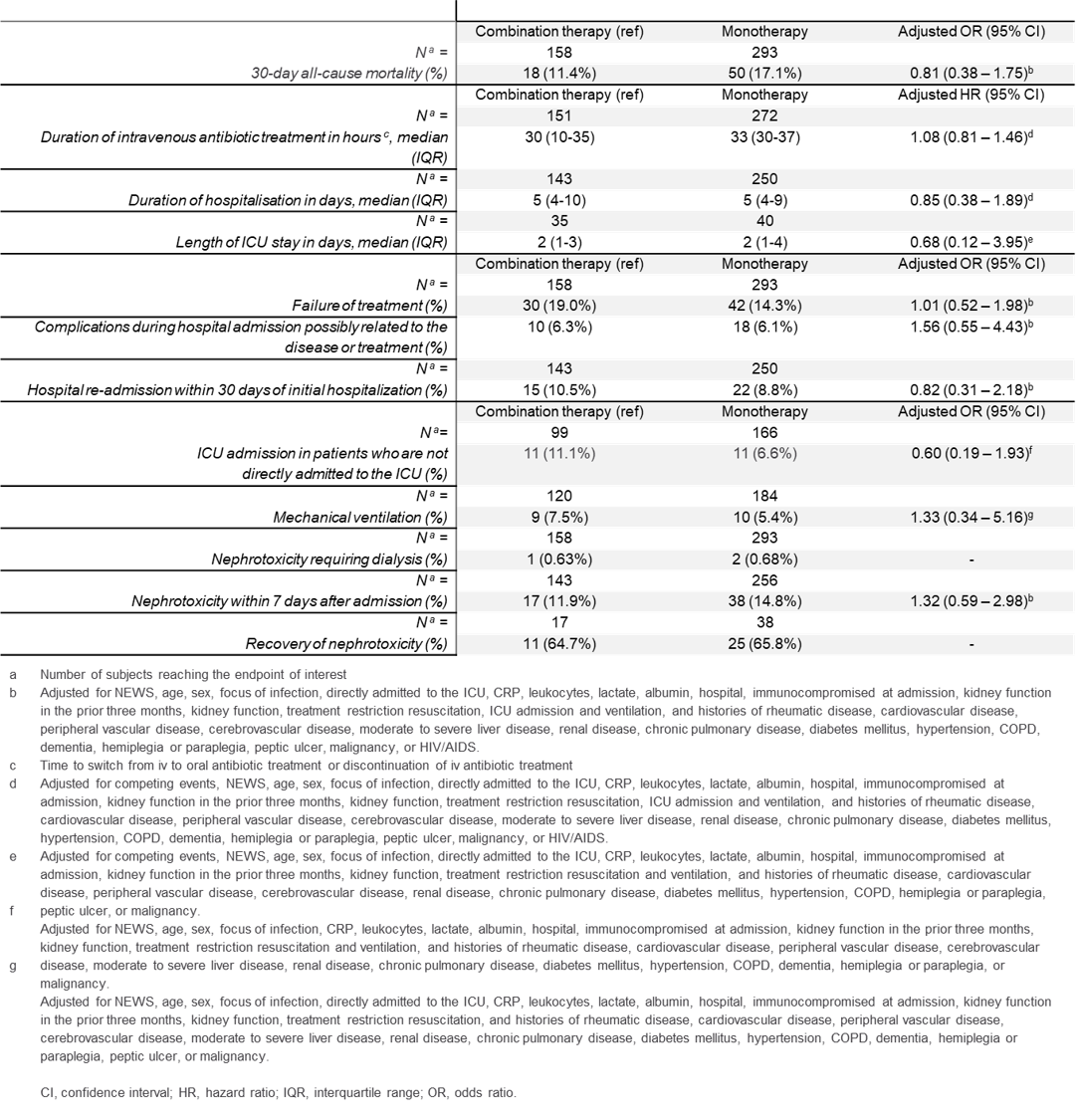
Failure to rescue a trial: lessons from an antibiotic therapy trial in the acute care setting
Sharon E.J.D. van den Eijnde ab*, Eva L. Koekenbier b, Paul D. van der Linden ab, Jan Jelrik Oosterheert c, Marc J.M. Bonten b and Cornelis H. van Werkhoven b on behalf of the SAGA study team
a Department of Pharmacy, Tergooi Medical Centre, Hilversum.
b Julius Centre for Health Sciences and Primary Care, University Medical Centre Utrecht.
c Department of Internal Medicine, University Medical Centre Utrecht.
* Correspondence: svandeneijnde@tergooi.nl.
Background and objective
In life-threatening conditions, such as sepsis, immediate initiation of antibiotic treatment is essential, which complicates individual randomisation. Cluster-randomized cross-over trials (CRXOs) are a feasible alternative based on previous experience in the acute care setting. The Short-course AminoGlycosides as Adjunctive treatment in adults with sepsis trial (SAGA) intended to determine whether cephalosporin monotherapy is non-inferior for mortality compared to cephalosporin combined with short-course aminoglycoside. Initially designed as CRXO trial, it was prematurely terminated due to continued low inclusion rates and a high non-compliance in the combination therapy arm despite efforts to resolve these issues. To learn from this experience, we evaluated the barriers for enrolment and compliance in this trial.
Methods
An online questionnaire was distributed to the nine participating hospitals in the SAGA study between January and March 2024. It addressed four themes: training and preparation, study procedures, deferred consent procedure, and medication prescribing. Questions regarding all these four themes were directed to the local research team (LRT), while the hospital staff received questions related to the medication prescribing theme only. Additionally, barriers to prescribing the randomized treatment were ranked by importance, from most important (= 1) to least important (= 6 or 7, depending on the number of barriers).
Results
In total, 23 questionnaires were completed by LRT members, and 54 questionnaires by hospital staff (Table 1). The LRTs were satisfied with the training provided by the central research team. However, implementing the inclusion and exclusion criteria, along with training the hospital staff and keeping them informed was perceived as challenging. Nearly half of the hospital staff in the combination therapy arm were hesitant to prescribe the randomized treatment due to concerns about potential side effects, a concern that was not reported by any staff in the monotherapy arm. The primary barrier for the hospital staff in the combination therapy arm was concerns about potential side effects (mean = 2.86, SD = 1.93), followed by supervisor recommending deviation (mean = 3.61, SD = 1.98). In the monotherapy arm, the main barrier was doctor or supervisor recommending deviation (mean = 3.00, SD = 1.75), followed by perceived lower effectiveness of the randomized treatment (mean = 3.83, SD = 1.65) (Table 2).
Conclusions
When implementing a CRXO in an acute care setting, continuous engagement and involvement of hospital staff throughout the study period is crucial. Pre-existing objections or negative experiences should be addressed through education meetings before the study begins.
Exploring anti-Xa activity and its correlation with C-Reactive Protein in Intensive Care Unit patients receiving a therapeutic dosage of nadroparin
Chaja N. Klein a*, Freek van Gorp a, Matthieu C.J. Bosman b, Frederikus A. Klok c and Lenneke E. M. Haas d
a Department of Clinical Pharmacy, Diakonessenhuis Hospital, Utrecht.
b Department of Clinical Chemistry, Hematology & Immunology, Diakonessenhuis Hospital, Utrecht.
c Department of Medicine – Thrombosis and Hemostasis, Leiden University Medical Center.
d Department of Intensive Care, Diakonessenhuis Hospital, Utrecht.
* Correspondence: cklein2@diakhuis.nl.
This study investigates the correlation between C-Reactive Protein (CRP) levels and anti-Xa activity in Intensive Care Unit (ICU) patients receiving therapeutic doses of nadroparin. Anti-Xa activity is commonly used to monitor the anticoagulant effect of low molecular weight heparins (LMWHs) like nadroparin. In a retrospective analysis of 44 ICU patients, CRP levels and anti-Xa peak activity were measured within 24 hours of each other. Spearman's correlation showed a significant, negative relationship between CRP and anti-Xa activity (Rs = -0.609, p < 0.001). This suggests that high CRP levels, indicative of inflammation, are associated with subtherapeutic anti-Xa activity. These findings align with prior studies linking inflammation, coagulation, and anticoagulation activity. However, limitations such as sample size and lack of antithrombin (AT) data warrant further research to confirm these results and explore the mechanisms underlying this correlation.
Model-informed precision dosing of eculizumab in patients with paroxysmal nocturnal hemoglobinuria
M. ter Avest a*, S.M.C. Langemeijer b, N.C.A.J. van de Kar c and R. ter Heine a
a Department of Pharmacy, Radboud University Medical Center, Nijmegen.
b Department of Hematology, Radboud University Medical Center, Nijmegen.
c Department of Pediatric Nephrology, Amalia Children’s Hospital. Radboud University Medical Center, Nijmegen.
* Correspondence: mendy.teravest@radboudumc.nl.
Background
Eculizumab is a lifesaving, yet expensive complement C5-inhibitor which is approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH). Eculizumab is dosed as a fixed dose, but previous studies in patients with atypical hemolytic uremic syndrome showed a large inter- and intraindividual variability in pharmacokinetics of eculizumab. Adequate therapy with eculizumab is necessary to prevent breakthrough hemolysis in patients with PNH. In case of supratherapeutic concentrations, the dosing interval could be prolonged to reduce cost and to improve patient-friendliness.
Objective
With this current study, we want to develop patient-friendly dosing strategies to improve treatment response at preferably reduced costs.
Methods
A prospective observational pharmacokinetic study was conducted. Pharmacokinetic sampling was performed at 1-3 separate occasions and both peak and trough samples were collected. Data were used to enrich a dataset used for our previous pharmacokinetic analysis of eculizumab in aHUS patients. Pharmacokinetic modelling was performed with non-linear mixed effects modelling. The final model was used to develop new dosing strategies with the use of Monte Carlo simulations in 1171 individuals.
Results
In total, 27 PNH patients were included in this study, with a total of 123 paired observations of time and free eculizumab serum concentrations. A two-compartment model with parallel linear and non-linear elimination best described the data. The addition of disease as a covariate on inter-occasion variability (IOV) resulted in a statically improved fit (P < 0.001, difference in objective function: −86). The estimates of the model were clearance 0.163 L/day (RSE 6%), volume of distribution 1 4.3 L (RSE 6%), volume of distribution 2 2.6 L (RSE 5%), Q 0.62 L/day, maximum rate (Vmax ) 25.6 mg/day (RSE 5%), plasma concentration for 50% of maximum rate (Km) 13.5 mg/L (RSE 38%).
A weight-based loading dose on day 1 of therapy followed by the maintenance dose on day 15 improved therapy. We predicted that 52.0% of the individuals reach effective complement blockade on day 7 of treatment with the standard loading dose, compared to 99.9% of the patients with the new loading dose. Drug costs are comparable for both strategies. A therapeutic drug monitoring based dosing strategy in the maintenance phase resulted in 16.2% dose reduction. Interval prolongation to three and four weeks was possible in 47.9% and 2.7% of the patients, respectively. A dose increase form 900 mg to 1200 mg was necessary in 3.2% of the patients.
Conclusions
An individualized, patient-friendly dosing strategy of eculizumab in PNH patients results in better treatment response at lower drug costs.
A systematic review on anti-Xa monitoring in the therapeutic use of Low-Molecular-Weight Heparins
T.C.C. Jaspers a*, P.C Remmelzwaal b, E.P.S. Weersink c, K. Meijer b and N. Khorsand d
a Department of Hospital Pharmacy, Elisabeth TweeSteden Hospital, Tilburg.
b Department of Haematology, University Medical Centre Groningen.
c Department of Hospital Pharmacy, University Medical Centre Amsterdam, location VUmc.
d Department of Hospital Pharmacy, OLVG, Amsterdam.
* Correspondence: t.jaspers@etz.nl.
Background
Low-molecular-weight heparins (LMWHs) are widely utilized, also in special patient populations (e.g. patients with renal impairment, pregnancy and obesity). Monitoring with anti-Xa measurements is often used but the effectiveness of anti-Xa-based dose adjustments remains uncertain.
Objective
This systematic review aimed to provide an informed assessment on the usefulness of anti-Xa monitoring and subsequent dose adjustments in these patients.
Methods
A systematic Pubmed search for records written in the English language and an abstract available was performed. Inclusion criteria were peer-reviewed articles concerning randomized controlled trials and cohort studies reporting anti-Xa levels in patients receiving therapeutic doses of subcutaneous administered LMWHs. The primary outcome was the incidence of clinical events, such as major bleeding and thromboembolic complications, compared between patients with and without anti-Xa monitoring. Secondary outcomes were (1) the correlation between anti-Xa levels and bleeding or thromboembolic complications and (2) the frequency of dose adjustments based on anti-Xa levels.
Results
A total of 43 studies, both randomized trials and observational studies, were included. All studies had a high risk of bias. Four studies indicated comparable rates of bleeding and thromboembolic complications between anti-Xa monitored and unmonitored patients. Measurement of anti-Xa levels was associated with frequent dose adjustments. The correlation between anti-Xa levels and both bleeding and thromboembolic complications was weak, if it was present at all.
Conclusions
This review highlights the limited evidence supporting the use of anti-Xa monitoring for dose adjustments in the therapeutic use of LMWHs. Therefore, we conclude that measuring anti-Xa levels is currently not justified. Only new, high-quality, preferably randomized studies could indicate otherwise.
Sessie 4: Procesoptimalisatie
Efficiency of clinical decision support system alerts on anticoagulants – a flashmob study in Dutch hospital pharmacies
J. Graafsma ab*, E.M.W. van de Garde cd, H.J. Derijks e, H.L. Hoge fg, J.E. Klopotowska hi, F. Karapinar-Çarkit jk and P.M.L.A. van den Bemt a; for the Dutch CDSS-efficiency study group
a Department of Clinical Pharmacy and Pharmacology, University Medical Center Groningen.
b Department of Pharmacy, Tjongerschans Hospital, Heerenveen.
c Department of Pharmacy, St. Antonius Hospital, Utrecht/Nieuwegein.
d Division Pharmacoepidemiology and Clinical Pharmacology, Utrecht University.
e Department of Pharmacy, Jeroen Bosch Hospital, Den Bosch.
f Department of Pharmacy, Wilhelmina Hospital, Assen.
g Gaston Medical, Eindhoven.
h Department of Medical Informatics Amsterdam UMC, University of Amsterdam.
i Amsterdam Public Health Institute, Amsterdam.
j Department of Clinical Pharmacy & Toxicology, Maastricht University Medical Center+.
k Department of Clinical Pharmacy, CARIM, Cardiovascular Research Institute Maastricht, Maastricht University.
Members of the Dutch CDSS-efficiency study group: Z. Brkic Admiraal de Ruyter Hospital Goes, C. van Kesteren Albert Schweitzer Hospital Dordrecht, I.M. Rigter Amsterdam UMC Amsterdam, D.C.W. Hiel Alrijne Hospital Leiden, K. Fasen Anna Hospital Geldrop, M. Duyvendak Antonius Hospital Sneek, A. Doppen Beatrixhospital Gorinchem, H. Fleuren Canisius Wilhelmina Hospital Nijmegen, M. Kerskes Catharina Hospital Eindhoven, K. Crommentuijn Dijklander Hospital Hoorn/Purmerend, T. Jaspers Elisabeth Tweesteden Hospital Tilburg, C.I. Vader Elkerliek Hospital Helmond, E. van Kan Gelre Hospital Apeldoorn, S. van Haga-Lakhi Groene Hart Hospital Gouda, A. Ledeboer Haaglanden Medical Center Den Haag, B.E. Bosma Haga Hospital Den Haag, M. Heijnen IJsselland Hospital Capelle aan den IJssel, D.J. Theunissen Ikazia Hospital Rotterdam, M. Tromp Isala Hospital Zwolle, A. Blenke Jeroen Bosch Hospital Den Bosch, N. van Rein Leiden University Medical Center Leiden, T.M. Leferink Maasstad Hospital Rotterdam, I. Brenkman Maxima Medical Center Eindhoven, E. Vrijkorte Medical Spectrum Twente Enschede, F. Karapinar-Çarkit Maastricht University Medical Center Maastricht, L. Knapen Nij Smellinge Hospital Drachten, N. Khorsand Onze Lieve Vrouwe Gasthuis East Amsterdam, P. Meijerhof- Jager Ommelander Hospital Scheemda, V. Bukkems Radboud Universty Medical Center Nijmegen, C. Langereis-Noll Rode Kruis Hospital Beverwijk, E. Geurkink Streekziekenhuis Koniging Beatrix Winterswijk, M. Al Fartousi Spaarne Gasthuis Haarlem, A. Harmsze St Antonius Hospital Utrecht/Nieuwegein, M.M. Ebbens St Jansdal Harderwijk, J. Korporaal-Heijman Tjongerschans Hospital Heerenveen, A.R. Dreijer Treant Zorggroep Hoogeveen, P van der Linden Tergooi Hilversum, P.M.L.A. van den Bemt University Medical Center Groningen Groningen, J. van Assen Van Weel Bethesda Hospital Dirksland, K. Shudofsky VieCuri Medical Center Venlo, N. Looman Wilhelmina Hospital Assen, E. Roobol Hospital Amstelland Amstelveen, R. Lammers Hospital Gelderse Vallei Ede and M. Liem- Moolenaar Hospital Rivierenland Tiel.
* Correspondence: j.graafsma@umcg.nl.
Background and objective
Clinical decision support systems (CDSS) aim to prevent adverse drug events by alerting prescribers for risky medication-related situations. Anticoagulants are frequently involved in these alerts. However, research shows that most CDSS generate an overload of not clinically relevant alerts, making the CDSS alerts inefficient. Whether this also applies for anticoagulant alerts is not well studied. Therefore, the main aim of this study was to investigate the efficiency of CDSS alerts for anticoagulants in Dutch hospital pharmacies. Secondary aims were to determine the efficiency of CDSS alerts for any medication, and the time needed to assess all alerts.
Methods
A multicenter, single-day, cross-sectional study was conducted using the flashmob design, measuring a small number of parameters during only one day. All Dutch hospital pharmacies (n = 69) were invited to participate. These pharmacies had to use CDSS that operate on both a national medication surveillance database and on self-developed clinical rules. Hospital pharmacists and pharmacy technicians collected data on the number and type of alerts and time needed for assessing these. The primary outcome was the CDSS alert efficiency for anticoagulants, defined as the percentage of anticoagulant alerts that led to an intervention. Secondary outcomes were the CDSS alert efficiency for any medication and the time expenditure evaluating all alerts. Descriptive data- analysis was used.
Results
Of the 69 hospital pharmacies invited, 42 (61%) participated. The efficiency of CDSS alerts for anticoagulants was 4.0% (interquartile range (IQR) 0.0-14.0%) for the alerts based on the national medication surveillance database and 14.3% (IQR 0.0-40.0%) for alerts based on clinical rules. For any medication, the efficiency was lower: 1.8% (IQR 0.8-8.3%) and 13.4% (IQR 2.0-23.5%i) respectively. The median time needed for assessing the relevance of all alerts was 2 (IQR 1:25-2:46) hours per day for pharmacists and 6 (IQR 3:11-8:13) hours per day for pharmacy technicians.
Conclusions
CDSS alerts efficiency is generally low, for both anticoagulants and any other medication, while the time investment is high. Therefore, optimization of CDSS alerts efficiency and impact is needed to reduce adverse drug events, for example by utilizing more specific patient data from the EHR and artificial intelligence.
Evaluation of Contamination Risk in Repeated Propofol Use with One-Way Valve: An In vitro Study
R. Van der Stelt, M.A. Molenaar, R. Goes, N. Prevoo, A. Vermeulen Windsant-van den Tweel, E. Kolwijck and M. Abdul Roda *
Jeroen Bosch Hospital, s-Hertogenbosch.
* Correspondence: m.abdul.roda@jbz.nl.
Background
Propofol, a widely used intravenous anesthetic, offers sustainability advantages over volatile anesthetics but has environmental drawbacks due to its production and incineration. Repeated use of propofol in the operating room can improve sustainability. However, improper handling increases the risk of contamination. A postoperative sepsis outbreak in 2010 at Haven Hospital, Rotterdam, was linked to the misuse of propofol vials, including repeated use of a single spike, poor hand hygiene, and reuse of vial remnants. The Dutch Health and Youth Care Inspectorate (IGJ) prohibits the reuse of propofol for multiple patients. Nationally, it is being investigated whether the use of a one-way valve in the perfusion line, in combination with protocols for preparation and hand hygiene, can reduce the risk of contamination. This would allow the use of propofol syringes for multiple patients.
Objective
This study evaluates whether propofol can become contaminated under simulated repeated use in an in vitro setting.
Methods
In this study, a setup was created where a prefilled 50 mL disposable syringe containing 2% propofol was connected to a 2-meter infusion line equipped with two one-way valves and a three-way stopcock (see Figures 1 and 2 for setup details). At the distal end of the infusion line, a 50 mL broth bag contaminated with Staphylococcus epidermidis, Staphylococcus aureus, and Escherichia coli (final concentration of 1000 cfu/mL) was attached to simulate a sepsis environment. Propofol was infused into the broth bag at a rate of 15 mL/hour for 45 minutes. After each 45-minute interval, the infusion line, including the one-way valve, was replaced between the propofol syringe and the three-way stopcock. This process was repeated three times per syringe. A total of 10 syringes were tested for bacterial contamination using blood culture media, simulating 30 patients. Approximately 10 mL of propofol remained in each syringe after three sessions. The syringes were sealed and sent to the microbiology department for sterility testing.
Results
No bacterial contamination was detected in any of the 10 tested propofol syringes; all contents remained sterile.
Conclusions
This study provides insights into the safe repeated use of propofol in a clinical setting with minimal environmental impact. Although past incidents indicate a contamination risk, the use of a one-way valve, combined with strict hand hygiene and adherence to protocols for preparation and administration, can significantly reduce this risk.
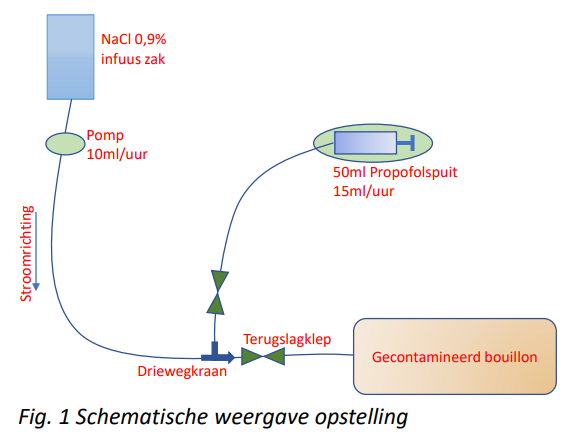

Verantwoording
De hier opgenomen abstracts vormen de mondelinge presentaties tijdens de Nederlandse Ziekenhuisfarmaciedagen op 28 en 29 november 2024 te Arnhem.
Referentie
Citeer als: Nederlandse Ziekenhuisfarmaciedagen 2024. Nederlands Platform voor Farmaceutisch Onderzoek. 2025;10:a1791.
DOI
https://www.knmp.nl/resolveuid/3770a2420f78446cb7ed0162dada39d8Open access

Reactie toevoegen